| Pages:
1
2 |
hissingnoise
International Hazard

Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Your amber liquid looks like my filtered cell-solution after a 30 hour run and a half-eaten cheap oiled graphite rod...
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Totally different animal though! 
Come to think of it.....years ago when I saved this dangerous amber beauty from disposal I thought that I shall likely never find a reasonable use for
it. In fact I mostly took it for the fancy bottle.....how wrong I was.
Exact science is a figment of imagination.......
|
|
|
Photonic
Harmless
Posts: 36
Registered: 29-5-2016
Member Is Offline
Mood: No Mood
|
|
For those investigating plating of platinum to titanium, or plating anything to titanium in general it is a fickle beast. One of the best references I
have found as of yet is the ASM Handbook Volume 5 - Surface Engineering.
It has some good information on plating solutions, recommended rinse methods, prep, etc.
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Some update on progress.....I tried to kill the 600C 4 coat anode by letting it run the cell to full conversion and beyond as far as could be
perceived. The poor thing was ticking along in the cell at 4,5-4,6V and 0,7-0,8A for three days, then chlorate depletion set in and current dropped to
0,3A. I let the torture continue for 7 more days in hopes of reaching the end for this Pt deposit. Aaaaaand, it does not die, nor wear, nor passivate!
I gave up on that one, dumped the cell contents to collecting flask and charged a fresh chlorate solution into the cell. At 4,4V I get 0,85-0,9A
current flowing, depending on how good the alligator clips manage to connect to the electrodes and the whole surface is working like brand new. This
is amazing....it means that the anode survived a full conversion and depleted cell conditions without a hint of passivation. That is a first, usually
I go through two anodes in a conversion run. The first one tends to die close to 3/4 th conversion and the second one ususally at the very end if
chlorate becomes depleted. But the 600C 4 coat survived without a damage and is currently processing a second conversion. I think this might be very
close to a sweet spot regarding the Pt deposition parameters. Let's see how far we can stretch this specimen 
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Methylene blue has arrived.....surely a lifetime supply for pennies:

Strong presence of perchlorate is indicated in the second conversion run of the 600C 4 coat anode
In fact the second conversion run is nearing a rather end stage and the anode shows no signs of degradation. It is running strong at 4,7V and 1A.
Defiantely this specimen is the most succesful anode attempt that I've managed to conjure so far.
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
The 600C 4 coat anode died at last......after accomplishing three full conversions of a total volume of 500ml concentrated sodium chlorate stock
solution. Likely even a bit more than 500ml as I added stock to the cell to replenish the losses during runs and it amounts definately more to 50ml
excess in total. Did not register the total amount of the additional chlorate stock, but then again I was just trying to find out how long the
electrode would last. The anode has been working constantly from the 5th of April until today, 6th of May. Minus the time it was off during recharging
the cell inbetween conversion batches, but that sums to a total of few hours at best.
A grand total of roughly 700h of operation in rather loosely controlled perchlorate cell conditions before It gave up the ghost. I think it is a
remarkable result for something with such a fragile and thin Pt coating. I must note that the first signs of degradation could be noticed at about
midway through the last conversion, so the specimen has been outstandingly rugged compared to previous ones that were deposited at a lower kiln
temperature.
I think it is time to execute the same deposition conditions on a larger substrate and try this with a scaled up version in the bigger cell. Shame the
Ti rod for the current collector has not yet arrived and I can not fabricate an operational setup, but the active surfaces can be prepared in advance
I shall percipitate the converted solution stock with KCl and measure the total yield. Or try with a smaller sample from the combined stock and
extrapolate for the total volume/mass. I really have no immediate need for KClO4, so might as well keep it in the solution form for any
further ideas I might come up with.
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
The Ti rods arrived, so I proceeded to fabricate a new anode stem for the "ubercell":
        
It should be at least grade 2 Ti by the looks and feels of it. Machines easily, is soft and pretty much ok to tap. I suspect the previous rod that I
used as anode stem was from grade 5 (alloyed Ti) and that material was tough as a coffin nail. Near to impossible for any tapping operations. By
extreme carefulness and a lot of prayer I succeeded, but it's not an experience that I would love to repeat. Well, let's see if it holds up in the
cell under anodic potential....
As for the active part of the anode I took a spent substrate from last time and cleaned it by sanding. Then deposited 5 coatings of Pt to it at 600C
in the lab kiln. Inbetween every coating the anode was wiped clean with a moist paper and then with alcohol until no more loose Pt could be detected
on the paper towel. The first coating had the most content of loose Pt dust, but from the third one forward there was literally nothing that could be
wiped off:
           
The Ti plate was attached to the slit end of the stem by compression fitting. It is pretty "goodentoit", but removable upon request and against my
intrinsic suspicions this type of connection seemed to work without passivation in the cell. At least it did so with the previous anode stem.
Charged into "ubercell" (the main conversion run with 600C 5 coat anode begins 08.05.19):
   
[Edited on 8-5-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Zooo....the contrapulation has been fizzing away on my tabletop for about 5 days now. The anode stem is not corroding, the anode has not passivated
and conducts very nicely and we have indication of perchlorate in the cell liqour. Both tests, methylene blue solution and percipitation by KCl show
the presence of perchlorate:

I did upgrade to larger cathodes as the stumps I had in there did hinder the process to such degree that I could only get 4,5A through the cell at
best. This will not do in terms of time expenditure....it would take forever to convert that load in the cell. So in went the large Ti cathode plates
I use for chlorate runs. I also made special Ti "bolts" to secure the plates to stems and a key to fasten them:
      
That's more like it....10 amps and temperature around 50C. I try to keep it around this mark as it is a safe middle ground. No excessive heating, but
still reasonable productivity. The liqour is quite clean as can be seen, so nothing of contaminative character seems to have dissolved into the
electrolyte. Seems to work out this time...
  
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
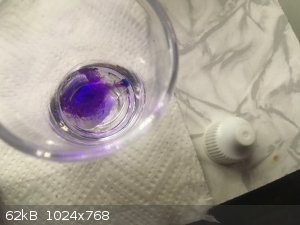
It is coming along very nicely. ClO4- content is growing steadily as recognized from precipitation test results. Ozone can be detected in
off gas flow, not much but it is clearly noticeable.
The titianium bolts securing cathode plates to the stems:

Anode edges can be seen to have lost conductivity (the darker rim around anode bottom part):

Not too bad though, the overall conductivity is very good:
The electrolyte is very clear and translucent. It has lost the greenish hypochlorite tinge, a telltale sign that the convesion is going well and has
reached a stage where perchlorate content has become prevalent.
There is offensive salt creep down the sides of the cell, clearly the seal at the top of the cell needs improvement, but this is more of a cosmetic
issue.
[Edited on 15-5-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
I did measure the KClO4 yield in the collected liqour from the small 150ml cell conversion runs and it came close to 0,9g/ml.....pretty
thick stuff
2 glass vials were cleaned, dried and weighed to establish tare mass. Then 2ml of collected cell liqour was inserted into one vial and weighed to
0,01g accuracy.
Density of the cell liqour was established as 1,5ml/cm3
6ml of saturated KCl soln. was added to the 2ml cell liqour sample to precipitate the product. 8ml of distilled water was added into the vial to
loosen up the cristalline layer and to enable it to separate from the liquid phase.
The liquid phase was removed with a syringe and needle to the maximum possible extent. Some of the precipitate was sucked along with the liquid. This
was injected into second vial an let settle. From second vial the liquid phase was again removed after the layer settled and then discarded. The main
precipitate was once again washed with 2ml of distilled water that was again removed with the syringe.
The vials were carefully microwaved to dry the precipitate and then weighed again.
The collective mass of preciptate from both vials summed as 1,8g.
I also measured the collective volume of cell liqour from the 150ml runs and it sums up to 610ml total weighing in at 913g d=1,49g/ml
The total expected yield when converted to KClO4 from this amount of liquor would be around 550g. Depending on how much losses occur at
purification.
That is the result of a month worth of electrolysis with the small cell running at an average guestimate of 0,6A.
If my very ragged calculations hold any water then this would amount to about 50,smt% current efficacy?
[Edited on 16-5-2019 by markx]
[Edited on 16-5-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
She's clearly cooked and done (08.05.19-24.05.19):
   
  
The cell liquid had obtained a deep clarity with almost a vitreous bluish tinge. High refractive index can be observed, characteristic of a
concentrated solution. Further torture is meaningless and shall only manifest itselt in electrolytic discharge of water and bringing the anode closer
to death.
I carefully suctioned the contents of the cell into a holding canister and discarded about 50ml that contained the slimy haze at the cell bottom.
The contents of the cell weigh in at 3672g by a volume of 2,5l. I visually assess that about 75-80% of anode surface is still active, so it should be
good for one further conversion of same magnitude.
Lets find out:
  
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
The second conversion is done.....so is the anode. I let it cook until it passivated completely at the maximum of 5,4V from the power supply. Also the
thermometer coupling disintegrated where the threading used to be for mounting it to the cell cover. The plastic electrode couplings also seem to turn
into slime. Nothing short of PTFE or PE is going to hold up very long in them fumes. But it finished 2 "ubercell" conversions with one anode, totaling
guesstimatively above 4kg of KClO4 and that is not too shabby at all!
It is time for an overhaul regarding the couplings and this time I aim for full PTFE set. Also a more effective seal for the cell cover is needed. The
makeshift plumber tape wound contraption is not really tight and has considerable salt creep+ some of the gas leaks out. Luckily it does seal against
the mist, but evident improvement is needed.
The second conversion was done with the contaminated sodium chlorate batch from first attempt that did not want to form any perchlorate with the old
(grade 5?) Ti anode stem. In fact it had formed some perchlorate after all as it turned out. It was faintly indicated with the methylene blue test,
but not enough to be indicated by the precipitation test with KCl. By the time I was running that batch, I had no methylene blue, so I could not
detect the faint presence of product. I filtered the goop out from the stored solution and gave it a try with the new anode setup. Worked beautifully
the second time and it was quicker than the first conversion.
So it seems my suspicions about the metallic contaminants from the dissolved anode stem hindering the conversion were unfounded. I guess the problem
was more likely of electrochemical nature (dissolution of the anode stem pulling down the anode potential enough to keep perchlorate formation at a
minimum?)....or the chlorate conversion was incomplete to begin with and it just took way longer than I had the patience to endure.
So far, so horrible.....but what the hell am I going to use the product for now?
I replaced the tired plastic couplings with a set I machined from PTFE stock.......should be way more durable:
   
The second conversion batch weighs in at 4111g at about 2,75l capacity:
 
A sample of 205g of the NaClO4 liqour was taken ans subjected to percipitation with saturated KCl solution:
  
The product was filtered on a glass frit :

[Edited on 3-6-2019 by markx]
[Edited on 3-6-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
The yield from the 205g (135ml) stock solution when converted into KClO4:
 
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
I did spin some visco based on the KClO4:
  
I am utterly unimpressed so far.....for something that required so much effort and fun to produce on my tabletop from scratch, it really performs no
wonders on it's own. But I guess that has more to do with the preparation (or the lack of) of the fuse composition.
I took a very simple approach: 75/25 KClO4/C (charcoal)
Ingredients were milled in electrical coffee grinder (obviously in separated turns) and mixed together in a bowl. 5-7% EtOH was added to the mix and
it was screened twice through a fine sieve (0,5mm) to yield a slightly granulated powder. It was allowed to dry completely in the sun for 2 hours and
then spun into fuse. The fuse was coated twice with diluted contact glue. A standard method I use also to coat BP based fuse. It yields a durable
flexible and waterproof product.
Well it does burn.....rather consistently, but :
really hard to light
not very agressive
still a lot of dross (not nearly overoxydised enough to burn away the threads)
I mean sure it was a halfbaked approach, but I expected at least some kind of wow effect
At least it does not stink of sulfur....
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
I also tried 87/13 and 65/35 ratios KClO4/C respectively. These do not burn at all or do so very slowly and tend to go out by their own.
So the workable range for visco I guess is at about 75-80/25-20.....
Still I am amazed at how feebly the perchlorate performs as an oxydizer....at least in a mix with just carbon. Chlorate in the same mixture is
exceedingly energetic and violent, even when the mixture is coarse and not well homogenized. But the sensitivity ruins the noble cause.
Perhaps if the the ingredients were ball milled to incorporate them into each other more thoroughly as can be done with BP based formulations. But I'm
not eager to try it with perchlorate, to be honest. Although it seems to be quite impossible to ignite the perch/C mix with a hammer blow, but still a
very imminent danger of a mishap is there.
Exact science is a figment of imagination.......
|
|
|
yobbo II
National Hazard
Posts: 762
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
Perchlorate is much more stable than chlorate
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Indeed it is....but I clearly did not expect the difference to be that drastic.
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Jeez....the simple K perchlorate+reducer mixtures seem so feeble and anemic because of the high decomposition temperature of the potassium salt that I
really see no possibility to use them straight as a core filling for visco. Also the usual accelerators for decomposition seem not to work on KClO4.
Interestingly a chinese patent ( CN101337855A ) describes a more complex mixture that overcomes the problem of high activation energy and slow
decomposition kinetics of KClO4.
Namely with the addition of 2-8 parts of K nitrate, 5-8 parts of Ca sulfate and up to 8 parts Ca carbonate as accelerants. Seems worth a try as I
tend to have all of these components at hand.
Exact science is a figment of imagination.......
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
I finally managed to devise a way to make the perchlorate work effectively in fuse formulations: the key is in preparation of the mix. It absolutely
needs a very intimate contact with the fuel, just like black powder based formulations.
There are two major ways to accomplish this in an amateur setting:
a) (ball) milling the mix- not viable for perchlorate formulations (at least my selfpreservation instinct does not allow it)
b) impregnating fuel by a perchlorate rich solution to form crystals in and around the fuel particles
I chose the second option. I made a small 2g sample of 75/25 KClO4/C, but this time I changed the preparation process to try out a "vaccum assisted
preparation" idea:
Ingredients were weighed into a small glass dish and 0,5ml of azeotropic EtOH +1ml water was added. The alcohol serves the purpose of lowering surface
tension to yield better wetting effect of carbon.
The ingredients were mixed with a spoon until uniform (a few minutes) and compacted into a solid cake. The consistency of the mix was like damp dirt
so it stuck together nicely.
After that the sample was placed into a vaccum chamber and evacuated for 30 minutes at room temperature (20C). Then the sample was taken out, the cake
broken up into loose rubble and put back into the chamber. Another session of 30 minutes under reduced pressure and slight warming at 40C yielded a
semi dry granulated product. It was still damp, but good enough to test for burning speed.
A small lump of the prepared mix was collected onto a stainless spoon and ignited with a butane torch: the sample burned with satisfactory violence
and was energetic enough to flying the burning lump off the spoon. Definitely the uniformity and vigor of the burn has been improved by the vaccum
assisted preparation process.
The slightly damp mix was granulated by forcing through a fine sieve and let completely dry at room temperature and atmospheric pressure (24h).
The sample was spun into a lenght of fuse after it had been dried:
 
It works just beautifully now! A nice hot, energetic burn that proceeds at an even pace with a sharp long flame jet and a powerful hiss. I'm very
satisfied with the outcome.
  
[Edited on 20-7-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
Raniphad
Harmless
Posts: 2
Registered: 25-7-2019
Member Is Offline
|
|
Great Information.
|
|
|
underground
National Hazard
Posts: 702
Registered: 10-10-2013
Location: Europe
Member Is Offline
|
|
Markx i got an MMO electrode and i want to spot weld to it a titanium bar for delivering the current. I have not any spot welder tho and i was
thinking to use some electrolytic caps in parallel, charge them, and spot weld it. What do you think, is it going to work ? I have seen some videos
that some doing it with a super-capacitor.
[Edited on 7-8-2019 by underground]
|
|
|
markx
National Hazard
Posts: 646
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Quote: Originally posted by underground  | Markx i got an MMO electrode and i want to spot weld to it a titanium bar for delivering the current. I have not any spot welder tho and i was
thinking to use some electrolytic caps in parallel, charge them, and spot weld it. What do you think, is it going to work ? I have seen some videos
that some doing it with a super-capacitor.
[Edited on 7-8-2019 by underground] |
Given enough capacity it will likely work, but I would think it takes a "bit" of tinkering around to get the parameters just right.
You could also try the compression fitting method I used with my perchlorate anode. Just squeeze the MMO mesh into a slot on the anode
stem....additionally one could compress it further with a Ti bolt e.g. It worked just fine without a bolt for me, despite my initial fear that the
connection spot would passivate in minutes rendering the system incapacitated. If such a connection will hold up in perchlorate cell conditions, then
I would believe it works well also in chlorate setups.
I is quite easy and offers the ability to be able to change the active part of the anode if needed. A spot welded system is permanent and can nod be
dissassembled.
Exact science is a figment of imagination.......
|
|
|
| Pages:
1
2 |









