nimgoldman
Hazard to Others

Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
Removing KOH from K-GHB
I synthesized some potassium gamma-hydroxybutyrate (K-GHB) by refluxing gamma-butyrolactone (GBL) with KOH in 80% ethanol (the idea was taken from Rhodium archive on Erowid, which is based on one of the cited articles where anhydrous ethanol was used as a solvent).
Unfortunately, I used little bit too much KOH and now I am stuck with the workup.
My first idea was to remove the solvent (ethanol + water), dry the solids and then wash them with acetone. Unfortunately, KOH does not really dissolve
in acetone and it reacts with it (aldol reaction).
Even the solvent removal and drying might be problematic as KOH is hygroscopic and caustic, so I am afraid it will damage the glass in the process. I
can reduce the b.p. by progressively adding ethanol to the mixture.
My other idea was to first neutralize KOH with some acid, then remove the solvent and perform washing.
At first I wanted to use acetic acid for neutralization, then wash out the potassium acetate with acetone. No luck, the acetate salt is not soluble in
acetone.
From the table on page 3 in this document, I know K-GHB is insoluble in: acetone, chloroform, ether, hexane. These solvents are candidate for washing.
Hexane is nonpolar so it won't help, but chloroform might be replaced by DCM which is kind of polar and might be able to dissolve potassium acetate or
other potassium salt.
I can use different acids too, such as hydrochloric, phosphoric, citric, tartaric, oxalic. One concern is of course the neutralization reaction not
harming the product (K-GHB).
Note: I am aware the said compound is a scheduled psychoactive drug. I am writing a harm-reduction article about the synthesis, since most
information I found on the internet about the topic is inaccurate to downright harmful. Most "recipes" I found would lead to incomplete reaction or
residual base making the solution too caustic. I am looking for a simple to follow purification technique for Na-GHB and K-GHB, the most common GHB
salts.
|
|
|
DavidJR
National Hazard
Posts: 908
Registered: 1-1-2018
Location: Scotland
Member Is Offline
Mood: Tired
|
|
Acidify with HCl to produce the free acid, distill under vacuum, carefully neutralise the free acid with KOH.
edit: or just tolerate having some harmless salt as an impurity
[Edited on 12-4-2019 by DavidJR]
|
|
|
nimgoldman
Hazard to Others
Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
Quote: Originally posted by DavidJR  | | Acidify with HCl to produce the free acid, distill under vacuum, carefully neutralise the free acid with KOH |
I should have noted the product should be K-GHB in pure powder form (kept in dry conditions ofc). So even distilling the acid and adding KOH carefully
will still yield a mixture of unreacted GBL, K-GHB and KOH and maybe some byproducts and impurities.
The free acid is unfortunately quite unstable and tends to cyclize to lactone, which has a high boiling point. I prefer to extract it with DCM.
So far the only way to work up K-GHB I found is with anhydrous ethanol and anhydrous ether and I have not even found a way to recrystallize it (it
does not crystallize from ethanol like Na-GHB). So maybe it will really be worth to do the step back and re-do almost entire synthesis:
- acidify
- extract GBL with DCM
- rotavap off DCM
- dry GBL with anh. Na2SO4
- distill GBL (fractional, vacuum, 2x)
- prepare NaOH in anh. EtOH
- slowly add GBL, react for 2h
- filter Na-GHB precipitate
- wash with anh. acetone
- dry
- recrystallize from boiling EtOH (2x)
- dry
It's a lot of work but better have pure Na-GHB than impure K-GHB. One of my goals is to build up a concentration/density table for common GHB salts
solutions so one can look up and estimate concentration knowing just density of the solution (provided there are no other salts and not an excess of
base, which will be tested separately).
One more idea is maybe to crash K-GHB from the reaction mixture with acetone. But given K-GHB's immense solubility in water, it might not work.
[Edited on 13-4-2019 by nimgoldman]
|
|
|
bipolar
Harmless
Posts: 24
Registered: 24-3-2019
Member Is Offline
|
|
I would probably try to kill excess of KOH with citric acid, evaporate solvent from your mixture (at the end adding some IPA to remove traces of water
azeotropically), dissolve solid residue in dry methanol at boiling, do a hot filtration from (presumably) insoluble potassium citrates that were
formed. Then chill in the freezer and hope for your product to crystallize out of MeOH solution.
This would work for Na-GHB and NaOH, not sure about potassium salts (different solubility).
If K-GHB won't come out of methanol solution, you could evaporate most of the solvent and see if it would crystallize now. If it doesn't, then maybe
evaporate even more solvent and dilute the residue with dry acetone; or at this point finally give up on potassium salt. 
[Edited on 13-4-2019 by bipolar]
|
|
|
Metacelsus
International Hazard
Posts: 2542
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
If you have any GBL left over, you could add just enough to react with the excess KOH.
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Do the reaction in water with excess GBL at boiling temperature . The excess GBL will boil away with the water. No need for ethanol. If you need dry
powder carefully heat at a bit above 100 degrees or dry under vacuum. When humidity is around zero it will dry even at room temperature.
[Edited on 13-4-2019 by Tsjerk]
|
|
|
nimgoldman
Hazard to Others
Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
Unfortunately I don't have any left (will make some later).
This is what did originally to bring pH down - but I found in one case the pH readings did not responded accordingly. I had a pH of 10.5, added 50 mL
GBL and the pH was 9.8, then added GBL in 10-50 mL increments pH was still around 9.7-9.8 ...
I then found there was too little water in the solution and just adding some water changed the pH reading drammatically (it lowered as expected).
Then I found the reaction performs badly in water and high-grade ethanol works much better.
I decided to do finish the reaction by reducing the amount of water and adding ethanol to reach over 60% concentration, refluxed the solution with
little excess KOH and then hoped to recrystallize only to realize K-GHB does not crystallize from alcohols.
I tried methanol and ethanol. Only Na-GHB crystallizes and only from ethanol (even 95% works, while K-GHB stays dissolved even in absolute ethanol).
So I finally given up on K-GHB, re-acidified the reaction mixture with HCl and plan to extract GBL (with DCM), re-distill it and do the reaction again
but this time in absolute ethanol, because now Na-GHB will simply precipitate and be recovered by filtration. Recrystallization in EtOH is also easy
and will remove both unreacted GBL and NaOH.
|
|
|
advanced warning
Harmless
Posts: 12
Registered: 6-5-2019
Member Is Offline
|
|
| Quote: Originally posted by DavidJR | Acidify with HCl to produce the free acid, distill under vacuum, carefully neutralise the free acid with KOH.
edit: or just tolerate having some harmless salt as an impurity
[Edited on 12-4-2019 by DavidJR] |
HCl wouldn't work, as the boiling point is significantly lower than GBL's. You'd recover mostly HCl and water before the distillation flask was choked
with precipitate.
Concentrated H2SO4 would eliminate this due to the high boiling point, and probably would let you distill away all the lactone. It's a fischer
esterification, and you're shifting the equilibrium to the lactone by boiling it away.
If you have pure GBL, why reflux with ethanol and KOH rather than aqueous KOH? The product is the same, and you can simply evaporate the water (and
unreacted lactone) to yield the salt.
It's base-promoted hydrolysis. People have been using the same reaction to make soap for a very long time. You're over-complicating it.
Beyond that, once dissolved in an aqueous solution (like gastric fluid) the salt will return to it's equilibrium between the cyclic and aliphatic
form, especially considering the pH of the stomach. As in, if you take the dry salt and dissolve it in water, and evaporate the water, and repeat this
over and over, you will be left with eventually nothing*** as the lactone form is present whenever the salt is dissolved in water.
You mostly want to to avoid pH higher than around 9 or 10 to prevent polymerization of the lactone, but even then the resulting polymer would be
bioresorbable and not worth worrying about.
Further reading:
https://www.mckendree.edu/academics/scholars/issue8/shemwell...
***edit:
You would not be left with "nothing" as there is ionic potassium or sodium in the solution as well. I find it hard to believe that you would be left
with a solid hydroxide, but my understanding of the literature is that a solution of GHB exists in an equilibrium with it's lactone and aliphatic
form, and the lactone is volatile. Therefore, some appreciable fraction of the lactone would surely be lost as the solvent is evaporated. Perhaps an
insignificant fraction, but I merely wish to draw attention to the implications of this equilibrium.
[Edited on 7-5-2019 by advanced warning]
|
|
|
nimgoldman
Hazard to Others
Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
On the Erowid GHB Synthesis FAQ V1.5, I read:
| Quote: | | All published preparations of GHB, or more correctly Na-GHB, refluxes butyrolactone with sodium hydroxide in various solvents, usually alcohol in
combination with water. |
The only synthesis done in just water provided poor yields and the water was still very basic, even though equimolar amount of lactone and base were
added.
The author (Rhodium) suggested the synthesis works much better in aqueous ethanol even "as low as" 40%.
This was my case. Even after refluxing for 2 hours in water, the pH was 10 and stayed at around 9 even after adding lots of GBL. So the problem was
not in GBL being impure (I checked the purity by monitoring still head temperature during vacuum fractional distillation and by checking the density -
both corresponded to GBL) but rather the reaction not being driven to completion.
The added lactone skews the final Na-GHB salt concentration and one have to fully dry it only to redissolve to get exact concentration.
I've done the synthesis using anhydrous ethanol with great success, the product simply precipitates and I recovered it by filtration (this was Na-GHB
though).
I then tried 80% ethanol as a solvent but unfortunately the NaOH reacted with it causing yellowing of the alcohol. NaOH solubility is greatly reduced
in such a strong alcohol. I also don't have stirring means to keep the pellets moving around in the suspension while the reaction mixture refluxes -
so I moved on to experimenting with more dilute alcohol (so that NaOH will fully dissolve prior to addition of lactone).
I will try next time with something like 50-60% ethanol so that all NaOH will be dissolved, then check the reaction with pH paper and neutralize with
more lactone if needed (as I don't want any other salts in the solution and also avoid recrystallization, if possible, since this requires relatively
large amount of >95% EtOH).
I've given up on K-GHB as - even though an interesting salt - it is too hard to obtain in dry crystalline form.
Another reason for using ethanol is easier to remove the solvent. With water, it takes lots of heating to evaporate it to the point of salt crashing
out of solution. Aqueous ethanol is easier to distill off and concentrate the salt solution to the point of solidifying - I can then break the
soap-like Na-GHB, air dry and break even more, then repeat the process until dry powder is obtained. I can wash the powder with dry acetone or cold
dry ethanol, re-dry and finally make a solution.
If the Na-GHB will be pure enough, I hope to be able to make a concentration-density curve for Na-GHB aqueous solutions to estimate concentration from
samples for harm-reduction purposes.
Presenting the alcoholic synthesis is itself a harm reduction project as the published syntheses using just water seem to be dangerous, leaving
unreacted base in the solution, leading to burning or strong reactions from varying amounts of lactone precent.
[Edited on 7-5-2019 by nimgoldman]
[Edited on 7-5-2019 by nimgoldman]
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Butyrate is basic by itself. A pH of 9 /10 is close to neutral, but not completely neutral because butyrate pulls a proton from water in an
equilibrium with the protonated butanoic acid. No matter what you do, it will never become neutral. A pH of 9 is so close to neutral I would call it
neutral, there is only 10^-5 mol/liter OH-.
Just use 1.1 eq gbl to NaOH in water and boil down the solution quite a bit. No more excess gbl because it boils away and no more NaOH because of the
excess gbl. This reaction is quantitative.
|
|
|
Keras
National Hazard
Posts: 957
Registered: 20-8-2018
Location: (48, 2)
Member Is Offline
|
|
Did you try THF as a solvant? It is polar, and probably wouldn't dissolve K-GHB
[Edited on 7-5-2019 by Keras]
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I don't understand solvents being used besides water. This reaction is easy as hell. Throw it together, boil down and you will be left with a
quantitive reaction without side products.
I might be stupid but using ethanol or THF is more stupid.
|
|
|
morganbw
National Hazard
Posts: 561
Registered: 23-11-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Tsjerk | I don't understand solvents being used besides water. This reaction is easy as hell. Throw it together, boil down and you will be left with a
quantitive reaction without side products.
I might be stupid but using ethanol or THF is more stupid. |
I understand 100% and am in total agreement with you.
I do have to say, that I have in the past, and will surely in the future as well,
have taken some stupid, dead simple reactions and screwed them up big time.
Just saying, even the simple stuff, has been screwed up by me.
Normally, I do not mention this to the entire planet. I just live and learn.
[Edited on 5/7/2019 by morganbw]
|
|
|
nimgoldman
Hazard to Others
Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
| Quote: Originally posted by Tsjerk | | Just use 1.1 eq gbl to NaOH in water and boil down the solution quite a bit. No more excess gbl because it boils away and no more NaOH because of the
excess gbl. This reaction is quantitative. |
Okay but why most syntheses in academic papers use ethanol:water ?
On the GHB Synthesis FAQ I read:
| Quote: | | The reaction is equimolar, and the reaction is driven to the right more readily in a dilute ethanol solution than in a plain aqueous
soln. |
and
| Quote: | | the method works for ethanol solutions as dilute as 40% |
This suggests that the reaction proceeds poorly in ethanol of concentration below 40%.
And then I read:
| Quote: | | When the pH comes down to 7, stop the heating. This normally occurs within an hour, probably after just half an hour. If the pH isn't down to 7 after
an hour of refluxing, adjust the pH to neutral with dilute HCl or conc. citric acid. |
I've read in several sources that optimal pH of Na-GHB solution is 7.5. The pH of course depends on butyrate concentration and acidity of the
water itself (e.g. dissolved CO2).
Now all this does not make any sense if pure Na-GHB is itself more basic. Adding acid will only produce lactone and an acid salt (unwanted
impurities).
Using molar excess of GBL would solve the problem of making crude solution, but I am not after Na-GHB solution, but a pure salt. I want to
write about preparation of Na-GHB powder in low tech setup, then about making solutions of it of exact concentrations (using scale and volumetric
flask) and measure density of Na-GHB for various concentrations to plot a curve. Any excess GBL present will of course skew the results - it has a
very high boiling point so it won't be possible to just "boil it out" (or do you think it will be carried off with water vapour?); needless to say
decomposition products appear in GBL at temperatures above 130 °C (at least from my observations).
[Edited on 8-5-2019 by nimgoldman]
|
|
|
Amos
International Hazard
Posts: 1406
Registered: 25-3-2014
Location: Yes
Member Is Offline
Mood: No
|
|
why not just add a bit of a nice food-safe acid like citric or ascorbic to bring pH down to neutral? Potassium citrate would, if anything, help the
taste.
[Edited on 5-8-2019 by Amos]
|
|
|
Keras
National Hazard
Posts: 957
Registered: 20-8-2018
Location: (48, 2)
Member Is Offline
|
|
| Quote: Originally posted by Tsjerk | I don't understand solvents being used besides water. This reaction is easy as hell. Throw it together, boil down and you will be left with a
quantitive reaction without side products.
I might be stupid but using ethanol or THF is more stupid. |
Well, I never attempted the reaction myself, so it was just a wild guess based on the previous post. If the reaction shall advance to quantitative
output in water, then yeah, go ahead with it. 
|
|
|
advanced warning
Harmless
Posts: 12
Registered: 6-5-2019
Member Is Offline
|
|
| Quote: Originally posted by Keras | | Quote: Originally posted by Tsjerk | I don't understand solvents being used besides water. This reaction is easy as hell. Throw it together, boil down and you will be left with a
quantitive reaction without side products.
I might be stupid but using ethanol or THF is more stupid. |
Well, I never attempted the reaction myself, so it was just a wild guess based on the previous post. If the reaction shall advance to quantitative
output in water, then yeah, go ahead with it.
|
Here is a patent application for a batch-process saponification of GBL to NaGHB with 95 to 99% reactant conversion. It uses equimolar concentrations
of GBL and NaOH in an aqueous solution.
https://patents.google.com/patent/WO2009129350A2/en
The purpose of ethanol as a solvent "allows product to be isolated as a solid from the reaction mixture without an additional recrystallization step".
It simplifies industrial-scale production by reducing residence time. The alternative would be to boil massive amounts of water to yield the
crystalline product. This would use a lot of energy, and reduce throughput of the factory. It's more cost efficient, industrially, to skip this step
and recrystallize from ethanol.
The only reason I could ever imagine a small scale chemist would use this method is if they fail to understand the reaction, and just copy someone
else's synthesis. Yes, GHB is produced industrially with ethanol, but the reason is because the reactants are incredibly cheap, and it means they can
do the reaction more quickly.
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
| Quote: Originally posted by nimgoldman | | Quote: Originally posted by Tsjerk |
Okay but why most syntheses in academic papers use ethanol:water ?
On the GHB Synthesis FAQ I read:
| Quote: | | The reaction is equimolar, and the reaction is driven to the right more readily in a dilute ethanol solution than in a plain aqueous
soln. |
and
| Quote: | | the method works for ethanol solutions as dilute as 40% |
This suggests that the reaction proceeds poorly in ethanol of concentration below 40%.
And then I read:
| Quote: | | When the pH comes down to 7, stop the heating. This normally occurs within an hour, probably after just half an hour. If the pH isn't down to 7 after
an hour of refluxing, adjust the pH to neutral with dilute HCl or conc. citric acid. |
I've read in several sources that optimal pH of Na-GHB solution is 7.5. The pH of course depends on butyrate concentration and acidity of the
water itself (e.g. dissolved CO2).
Now all this does not make any sense if pure Na-GHB is itself more basic. Adding acid will only produce lactone and an acid salt (unwanted
impurities).
Using molar excess of GBL would solve the problem of making crude solution, but I am not after Na-GHB solution, but a pure salt. I want to
write about preparation of Na-GHB powder in low tech setup, then about making solutions of it of exact concentrations (using scale and volumetric
flask) and measure density of Na-GHB for various concentrations to plot a curve. Any excess GBL present will of course skew the results - it has a
very high boiling point so it won't be possible to just "boil it out" (or do you think it will be carried off with water vapour?); needless to say
decomposition products appear in GBL at temperatures above 130 °C (at least from my observations).
[Edited on 8-5-2019 by nimgoldman] |
|
1 M of the butyrate would have a pH 9,3, with a pKa of 4,7. Almost neutral I would say. Concentrated solutions g(around 5M) go towards a pH of 10
What you forget in the comparison between water / ethanol reactions is that a boiling solution of Na-GHB in water is way hotter than a solution
containing ethanol. In my experience the reaction is complete after five minutes boiling, or when mixing cold by the time the reaction reaches boiling
temp. And yes, the surplus of GBL boils away with the water, via a sort of steam distillation. Just use enough water.
GBL does not decompose at 130 degrees. At atmospheric pressure, distillation is a standard technique to purify GBL
If you want to make dry powder I can imaging using 99% ethanol, then you only have to filter. But in water also works, it dries quite easily when in a
dry environment, just put it over CaCl2 for a week or so.
Last advice ; if you want this compound to be pure, use an excess GBL in water and boil down, ignore the pH. If you set it to seven you only push it
back towards GBL. Boil down to 1.5/1.75 time the volume of the original GBL, when this cools it will crystallize to a solid mass. Powder and dry over
calcium chloride or similar.
[Edited on 8-5-2019 by Tsjerk]
|
|
|
nimgoldman
Hazard to Others
Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
Okay I think it is clear now. Up to now I though pH 9-10 will burn tissue.. I have to learn more about strong and weak bases and salts and how it
affects tissue.
I tried different reaction setups and 99% ethanol is the fastest and producest perfect product - the precipitated powder is already very fine and no
further processing is needed.
On the other hand, one has to use relatively large amount of ethanol, something like 800 mL per 100 g of lactone. Lots if it is wasted during drying.
For plain water, it is very hard to boil off the water to get powder and the whole drying and powdering process is very tedious - I have to repeatedly
more the material between food dehydrator and mortar to break the lumps to increase surface area and dry it more. It takes whole day until the powder
is sufficiently fine and dry.
I cannot use the lumps because the trapped water adds weight and skews my density measurements.
Yeah I think something like 50% ethanol will be way to go as it will dissolve the sodium hydroxide easily, won't react with it, the reaction time will
be lower and it will be easier to boil it off (lower temperature as you said).
I produce GBL from GABA and even after second fractional (vacuum) distillation, the GBL darkens a bit in the boiling flask. I am not sure what kind of
impurity that could be...
My last batch of Na-GHB is yellow because the ethanol used was too strong and reacted with sodium hydroxide. Now I need lots of pure ethanol to
recrystallize the Na-GHB it and remove the yellow stuff (aldehydes?).
Thanks for all the advice so far.
[Edited on 8-5-2019 by nimgoldman]
|
|
|
advanced warning
Harmless
Posts: 12
Registered: 6-5-2019
Member Is Offline
|
|
| Quote: Originally posted by nimgoldman |
For plain water, it is very hard to boil off the water to get powder and the whole drying and powdering process is very tedious - I have to repeatedly
more the material between food dehydrator and mortar to break the lumps to increase surface area and dry it more. It takes whole day until the powder
is sufficiently fine and dry.
I cannot use the lumps because the trapped water adds weight and skews my density measurements.
|
KGHB is very deliquescent. You can dry it in a sealed box with a desiccant. If you take an aqueous solution of KGHB and boil off the water, eventually
the boiling slows but some bubbles still evolve as the temperature increases. The water is gone once the temperature starts increasing, despite the
slight bubbling. You want to stop heating when the vigorous boiling stops. This is 150C or so. Further heating degrades the product, leading to a
yellow creamy solid (like ice cream) rather than rock hard white crystals (visually like coconut oil).
Pour off the molten KGHB and allow to cool. You are left with incredibly hard white crystals. It solidifies as a block. You have to use a hammer to
break it up. It takes a very long time to dissolve in water. I'd recommend doing this only one time so you have the experience, but in future runs
it's wiser to not let it cool fully. Instead, let it cool below 100C and add water to get your desired concentration.
edit:
Perhaps your problems are due to the nature of the diazotization reaction? Reacting an aliphatic primary amine (like GABA) with nitrous acid generates
loads of products. The carbocation is very unstable, and will decompose into alkyl halides, alkenes, rearrange to a secondary alcohols, etc.
You would have a mix of compounds and a very tough time isolating them, I would imagine. Maybe I'm wrong, and maybe you've analyzed your end products
and they have acceptable purity.
I can't help but think it would be more worthwhile pursuing a different pathway. Succinic acid is readily available, and only needs to be hydrogenated
to yield GBL. This is done industrially with a metal catalyst, like cupric oxide, copper chromite, zinc oxide, Pd/C. This is perhaps easier said than
done, as most patents are focused on industrial applications. Generally vapor-phase hydrogenation of succinic anhydride or maleic acid at around 200C.
Very few are batch process, and every instance that I've seen of liquid phase hydrogenation is done at pressures of 20 bar or more. Here are examples
of high quality patents:
https://patents.google.com/patent/US6297389B1/en
https://patents.google.com/patent/US5055599A/en
Perhaps it could be done with lab glassware with more conventional catalysts. I'm unsure why liquid phase hydrogenation of succinic acid couldn't
occur at atmospheric pressure.
[Edited on 9-5-2019 by advanced warning]
|
|
|
oberkarteufel
Harmless
Posts: 47
Registered: 11-12-2018
Member Is Offline
Mood: Mayonesium sulfate
|
|
About the pathaway starting from succinic acid:
Ha, no further than 3 days ago I found in my Vogel a synthesis of (IIRC) homophthalic acid, with an intheresting intermediate - phthalide. It's
synthesis looks like this:
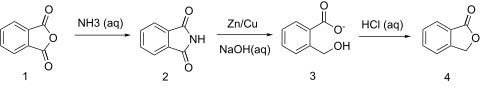
So I wondered, if such reaction is possible with succinic acid/anhydride/imide/choose your own starting point.
|
|
|
advanced warning
Harmless
Posts: 12
Registered: 6-5-2019
Member Is Offline
|
|
| Quote: Originally posted by oberkarteufel | About the pathaway starting from succinic acid:
Ha, no further than 3 days ago I found in my Vogel a synthesis of (IIRC) homophthalic acid, with an intheresting intermediate - phthalide. It's
synthesis looks like this:
So I wondered, if such reaction is possible with succinic acid/anhydride/imide/choose your own starting point. |
That's a very interesting synthesis you've posted. Apparently, yes, you can do the same thing with succinic acid. The reaction with ammonia, at least,
to form succinimide.
https://thosci.com/synthesis-of-suiccinimide/
Unrelated to the topic at hand, but from there, you can brominate the succinimide in an aqueous solution with NaOH to yield NBS. Potentially a very
useful pathway, from OTC chemicals.
I was interested more in the reduction of succinic acid to yield THF, but if the pathway you've posted holds up for succinic acid, it may be an easily
accessible option for GBL.
edit:
I can't imagine it would be all that hard to construct a suitable pressure vessel for containing such a reaction. The biggest issue would be
pressurizing a hydrogen atmosphere. I don't know how you could do this outside of an external tank. Perhaps you could generate hydrogen gas in some
sort of sealed container (like the NaOH + Al method). I'm imagining a pressure cooker type system with an attached pressure gauge. You could attach
some sort of ball valve and gas line to the reaction vessel, and pressurize it.
You'd still need some way to flush the system with nitrogen, or risk turning your steel reaction vessel into an IED. Nitrogen tanks are fairly cheap I
imagine. It seems fundamentally simple though. So long as you take appropriate precautions, you could easily hydrogenate effectively anything,
forever, in a batch-wise process. So long as you have a catalyst, of course.
Am I wrong in this assumption?
[Edited on 9-5-2019 by advanced warning]
|
|
|
nimgoldman
Hazard to Others
Posts: 304
Registered: 11-6-2018
Member Is Offline
|
|
Thanks for all the input.
The pathway from GABA is certainly not the higher yielding one but for my purposes it is good enough.
The purity of the product relies on purification of GBL by vacuum fractional distillation. I found two runs produce a product with almost constant
boiling point. I don't believe there are many side products with such high boiling point as GBL and soluble in both water and DCM.
Today I will test Na-GHB powder (synthesized in alcohol) for pH and if not too high (11 or more), I will just wash it with acetone, dry and make a
solution.
If the pH will be too high, I will recrystallize it from 95% ethanol.
As for alternative pathways, I found people also use THF (tetrahydrofuran) and there are many syntheses to go from THF to GBL.
The succinic acid route is also interesting and I might pursue it later (I have only limited time for this project). As for the pressure vessel, I
have a small "hydrothermal reactor", which is a PTFE container inside thick steel vessel with threaded cap - basically a fancy pipe bomb for high pressure
syntheses.
|
|
|
draculic acid69
International Hazard
Posts: 1371
Registered: 2-8-2018
Member Is Offline
|
|
Are you talking about LAH reduction or noble metal catalyst reduction of succinic acid?
|
|
|
advanced warning
Harmless
Posts: 12
Registered: 6-5-2019
Member Is Offline
|
|
Industrially it's generally directly reduced to GBL THF, but the other pathway linked is through succinimide.
This patent:
https://patents.google.com/patent/US2919282A/en
reveals that phthalimide can be reduced with various metals, such as copper, aluminum, and zinc with an alkali hydroxide. When reacted with acid, this
yields phthalide. The yields are reported to be around 90-95%.
This should be analogous to succinimide to GBL. The literature is lacking, but correct me if I'm wrong on this assumption.
Of course, this reaction may not be necessarily more practical than GABA diazotization, as copious amounts of NH3 gas would need to be safely vented.
|
|
|








