| Pages:
1
2 |
Jackson
Hazard to Others

Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood:  Happy about new glassware
|
|
molecule synth
Hi, I was wondering how I could potentially make this molecule
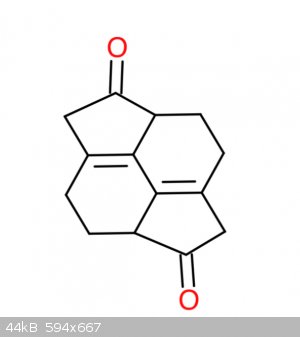
|
|
|
j_sum1
|
Thread Moved
23-9-2018 at 14:03 |
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Is there any particular reason why you have chosen this structure specifically?
Im working on a theoretical pathway but its tricky, its always good to look back at what others have done before and thus if you have an origin for
the compound i could look at how it was originally prepared.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Ok ive gotten thus far, ive yet to begin researching the preparation of the 3-hydroxy-2-(2-hydroxyethyl)cyclohexanone.
Your compound is insane, might i add.
The route goes like so.
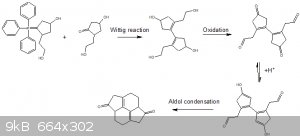
Step 1
It begins with a wittig reaction, the preparation of the ylide reagent would have to likely start with triphenylphosphine reacting with a alkyl halide
followed by addition of a strong base.
From what i understand, both of those hydroxides will not react at all with the phosphonium species or triphenylphosphine however they will be
deprotonated by the base.
If i am wrong then one could instead start with both hydroxides oxidized to there corestonding carbonyls (aldehyde and ketone) which could be
protected by an ethylene dioxide acetal/ketal dioxolane group.
Step 2
Then the hydroxides can be oxidized or any protecting groups cleaved.
Step 3
The next step is where there is a bit of a problem, it requires an enolation of that ketone in order for the aldol condensation to take place, the
major issue is that there is no selectivity for which side the proton is taken from, see below.
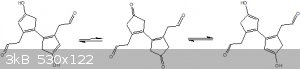
Now ive drawn up some other possible side products that could form from this aldol condensation, non of which seem sterically favorable, even if the
enol does form on the wrong side, the corresponding aldehyde cannot react with it as the chain is far too short in length.
I also doubt the aldehyde on the other side of the cyclopentene ring will reach over to react with it (sterically unfavorable).
This leaves intermolecular side reactions, for which i might propose a very high dilution within the reaction mixture to attempt to avoid such side
reactions.
With that enolation in mind, i suspect the yields for that condensation will be on the low side, although one might be able to recycle any unreacted
product.
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
My original molecule was even worse with cyclobutane rings instead of cyclopentane rings. I’m working on making shapes at a molecular level kind of
like Bucky balls.
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
2-(2-hydroxyethyl)cyclopentanone Could possibly be synthesized by a reaction of 2-bromocyclopentanone with 2-bromoethanol.
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
Wouldn't the wittig reaction give the alkene instead of the butadiene? Perhaps a pinacol coupling of two cyclopentanones followed by a double
elimination would work.
Edit:
The claimen condensation gives two extra double bonds you need to get rid of...
[Edited on 24-9-2018 by Sigmatropic]
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Would a pinacol coupling work with the 3-hydroxy 2-(2-hydroxy-methyl)-cyclopentan-1-one or would it need to be a different molecule?
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
@Jackson, how are you going to determine whether or not you get any product? What techniques do you have access too?
Cool synth!
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
I know some people at a university and I am working on getting access to a spectrometer.
|
|
|
Tsjerk
International Hazard
Posts: 3032
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Do you have a certain absorption spectrum you expect from this molecule?
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
the easiest way would be to reduce 1,5-pyracenedione (CAS no - 155143-87-6).Sadly I don't know how ,but I think the hydrogenation experts on this
forum(ex- nicodem , zed) would be able to help.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
@Sigmatropic
You are correct, I like the pinacol coupling elimination idea, so much so that ive included it in this new scheme.
I had a reason for proposing the aldol condensation that would not directly result in hydolosis to the beta alkene by instead picking it up with a
stong base such as sodium hydride. It was a late night though so ive decided that idea was far to insane and experimental to work.
I have made some massive modifications, although increased the number of steps quite significantly.
Im still not sure how one would go about preparing the cyclopentanone precursor but im thinking along the lines of a narzarov cyclization.
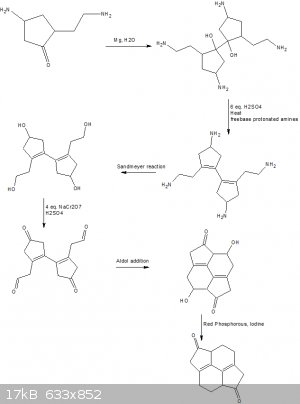
I have instead chosen to start with primary amines as place holders (protecting groups sorta) for our eventual Ketone/aldehydes.
Step #1
Pinacol coupling, where magnesium donates an electron to the ketone carbon forming a organo magnesium oxide, which proceeds to donate its second
electron to another mol of our cyclopentanone species, this organo magnesium species then hydolyses to give the corresponding tertiary vicinal diol.
I know very little about how to perform such a reaction in a practical setting but im hoping the beginning step is not water sensitive as it would be
ideal to avoid anhydrous conditions to prevent the formation of any imines.
Step #2
Simply acid calatysed elimination of the vicinal diol resulting in the formation of a diene in concordance with zaitsevs rule.
Step #3
The next step would be a classic sandmeyer reaction with sodium nitrite and HCl followed by hydrolosis of the diazonium cation.
Up to this point i was hoping to avoid anhydrous conditions to prevent the formation of imines, I still have no clear route to the precursor though so
this may change.
Step #4
Careful oxidation of the alcohols to yield ketones and aldehydes.
This step may be quite tricky, avoiding over oxidation of the primary alcohols with that many oxidation's taking place on the same molecule would be
troublesome.
It may instead be better to attemp using bismuth tribromide to selectively oxidize the alcohols.
I have posted this reference in the acetaldehyde thread but will post here too.
Attachment: lee2015.pdf (143kB)
This file has been downloaded 346 times
Step #5
The aldol Addition reaction where instead of eliminating a molecule of H2O as in a traditional aldol condensation, i would instead
propose reducing this alcohol to a saturated alkane.
Step #6
Reduction of the alcohol using red phosphorous and iodine which is self explanitory.
Im not sure how that beta ketone is going to affect the ability of the iodine to leave but i would surmise that the carbonyl carbon with its
electrophilic nature would not hinder the oxidation of the iodide by HI, thus this should work fine.
Now, Mr Jackson, as you can see this is no easy compound to prepare and is well beyond my own skills (and i atleast have some comprehension of the
undertaking).
You would be hard pressed to convince someone else to carry out such a multi step organic synthesis especially since we are already at 6 steps without
even considering preparing the precursor.
With that in mind, i have enjoyed thinking about this and do not want to discourage you from bringing such problems to the forum, even if it is purely
theoretical.
If you really are considering a project similar to this though, might i propose something a little less insane.
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
I was working on figuring out how to make the precursor. I think that a divinyl ketone with an amino group on one end could undergo a Nazarov
cyclization using something like TiCl4 to form a 4-amino-cyclopentanone. Then it could be reacted with a halogen like chlorine to form
4-amino-2-chloro-cyclopentanone. Then it could be reacted with 2-chloroethylamine to form 4-amino-2-(2-ethylamine)cyclopentanone.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Assured Fish  | Step #2
Simply acid calatysed elimination of the vicinal diol resulting in the formation of a diene in concordance with zaitsevs rule. |
It will undergo a pinacole-pinacolone rearrangement instead -https://en.wikipedia.org/wiki/Pinacol_rearrangement
| Quote: | Step #6
Reduction of the alcohol using red phosphorous and iodine which is self explanitory. |
HI/red P would take out
the double bonds too
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
CuReUS the pinacol rearrangement is an entirely different reaction, taking place on a vicinal diol, it required acidic conditions in order to
protonate one of the alcohols so it can leave as H2O. Given that the by product of the pinacol coupling is magnesium hydroxide, i doubt it would take
place without the operator making it take place.
https://en.wikipedia.org/wiki/Pinacol_rearrangement
I had not thought about HI adding across the double bond, good point.
I guess then my sodium hydride idea is looking more plausible.
Ive had trouble finding a way to get to the saturated alkyl ketone in an aldol fassion, surely its possible though?
Looking at the aldol condensation mechanism, it seems only logical that you could reduce the beta alcohol instead of hydrolysing it, is it possible
for an alcohol to react in an aldol condensation with an enol instead of classically reacting an aldehyde with an enol?
Edit: SHIT i didn't even think about H2SO4 acting as that proton source in the following elimination step. Well without another way
to eliminate those alcohols that doesn't require an acid of some kind, this route wont work.
[Edited on 26-9-2018 by Assured Fish]
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Maybe the alcohols could be converted to a different group and then be removed?
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Jackson that was exactly what i was thinking.
The obvious solution would be reacting the diol with HBr followed by elimination using sodium methoxide.
I also completely forgot that this was a thing, meaning the wittig coupling isnt entirely out of the question although i think given that it uses a
very strong neucleophile and those amines would be halogenated by any halogen we introduce, it would still be better to opt for the pinacol coupling.
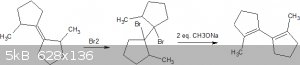
| Quote: |
Then it could be reacted with a halogen like chlorine to form 4-amino-2-chloro-cyclopentanone. Then it could be reacted with 2-chloroethylamine to
form 4-amino-2-(2-ethylamine)cyclopentanone.
|
This wont work however, as the halogenation could form on either side of the ketone meaning you would get 2 products, 1-chloro-2-amino-cyclopentanone
(undesired product) & 1-chloro-3-amino-cyclopentanone (desired product).
Separation of these 2 products would likely be a pain in the ass.
The second major issue is that halogens will react with the amine forming chloramines or bromoamines or whatever.
The third major issue is that you wont really be able to form an alkyl group from that halide without destroying other funtional groups.
The wurtz fittig reaction might maybe work but that reaction is outdated for the very reason of it being incompatible with alot of functional groups.
Either way it would be better to instead start with the alkyl chain in the right position prior to carrying out the cyclization.
Something like this perhaps:
|
|
|
Jackson
Hazard to Others
Posts: 189
Registered: 22-5-2018
Location: U S of A
Member Is Offline
Mood: Happy about new glassware
|
|
Could that precursor be made by reaction Propionaldehyde with sec-Butyl Magnesium Bromide to from 4-methyl-3-hexanol which could be dehydrated by CrO3
to form 4-methyl-3-hexanone. Then It could get nitrited to form 1,6-nitrite-4-methyl-3-hexanone. I dont know how to reduce the methyl group to a CH2
group but that would be the next step.
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
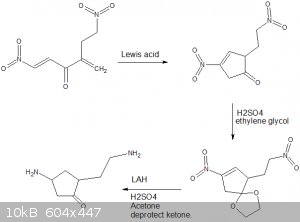
Another member brought this thread to my attention, and my thought was to try to construct those double bonds by a Hofmann elimination.
Retrosynthetically, if you "undo" the Hofmann elimination, you get something that looks like an iminium-driven aldol condensation product. That line
of thinking led me to this:
The sulfonation of naphthalene with oleum gives napthalene 1,5-disulfonic acid:
http://en.wikipedia.org/wiki/Armstrong's_acid
Alkaline hydrolysis gives 1,5-dihydroxynaphthalene:
http://en.wikipedia.org/wiki/1,5-dihydroxynaphthalene
According to this paper, acylation of 1,5-dimethoxynaphthalene preferably gives 4,8-disubstitution (when p-methylbenzoyl chloride is used):
https://ntrs.nasa.gov/archive/nasa/casi.ntrs.nasa.gov/200501...
So, without loss of sanity, we hypothesize the production of 4,8-diacetyl-1,5-methoxynaphthalene, at which point I will leave the published literature
and start speculating.
First, we protect the acetyl groups as their O-trimethylsilyl enol ethers, then we deprotect the methoxy groups using IBX.
The next step is a Birch reduction, which on phenols produces cyclohexenones. We expect the enol ethers will be left alone because they are
electron-rich and so will be less likely to react with e-. The product is compound (2).
Reaction of the product diketone with dimethylamine and acid produces a double directed-aldol condensation, and deprotection of the silyl ethers with
HF gives the compound (3).
The central double bond in (3) can be reduced with very high selectivity by diazane:
http://en.wikipedia.org/wiki/Reductions_with_diimide
Reduction of the double bond in (3) and methylation with MeI gives (4), which can then undergo a double Hofmann elimination to give the target
compound by Zaitsev's rule.
EDIT: I drew (3) wrong, you'd have to deprotect (3), then diimide.
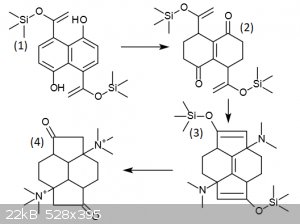
[Edited on 27-9-2018 by clearly_not_atara]
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
Let us not forget that the more stable isomer of the title compound is actually a double alpha,beta-unsaturated ketone (see attached scheme).
Several routes I drew up (not shown, because flawed) and the synthesis proposed by clearly_not_atara will preferably end up with an a,b-unsaturated
product instead trough virtue of the Ecb1 reaction (https://en.wikipedia.org/wiki/E1cB-elimination_reaction#Aldo...).
I doubt the title compound is accurately represented by the proposed stucture and as such attempts at synthesizing the title compound through
hypothetical routes are merely paper chemistry excersizes, which are interesting none the less.

[Edited on 27-9-2018 by Sigmatropic]
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Oh, there is that.
Maybe you can reduce the central bond selectively with diimide without touching the enol ethers, because diimide prefers symmetrical alkenes. Then the
elimination would give the right isomer, and deprotection of the TMS groups gives the correct product.
[Edited on 27-9-2018 by clearly_not_atara]
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
clearly_not_atara, the problem is you would end up with a different a,b-unsaturated ketone (see attached file).
Or am I missing something?
The stucture in the OP is defintetively only a kinetic product and most other syntheses will go right on to the stucture described earlier.
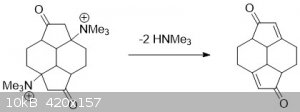
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
That position is blocked. See the picture. Deprotection of the silyl ethers in the final product will not cause rearrangement of the internal double
bonds by any mechanism I am aware of.
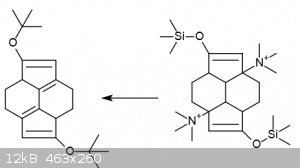
|
|
|
j_sum1
Administrator
Posts: 6336
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Ok. I don't think this is "Beginnings" any more. Even though the OP contains no citations and few references follow.
Nearly all of this goes straight over my head, but I love this kind of discussion. It is one of the things I love about this place.
If we had a name for this molecule it could be put in the thread title.
Back to OC.
|
|
|
j_sum1
|
Thread Moved
27-9-2018 at 16:20 |
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
I came up with a modified version of fish's route ,aka the fureus route 
the starting compound is available commercially ( CAS no - 73039-22-2)
1.converted to the lithium enolate followed by reaction with oxirane to give the ethyl alcohol
2.Protection of the alcohol with benzoyl chloride/Pyridine -https://www.synarchive.com/protecting-group/Alcohol_Benzoate
3.pinacol coupling with Mg,followed by double dehydration with POCl3/Pyridine ,avoiding the subsequent rearrangement -https://www.masterorganicchemistry.com/2015/04/28/eliminatio...
3.Simultaneous deprotection of the alcohol and vinyl ester to give the corresponding cyclopentenone alkyl alcohol
4.Tosylation of the alcohol followed by treatment with LDA to give the target compound via an intramolecular alkylation.

we could call it "synthesis of
hexahydro pyracenedione"
[Edited on 28-9-2018 by CuReUS]
|
|
|
| Pages:
1
2 |








