| Pages:
1
2
3 |
Assured Fish
Hazard to Others

Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Capsazepine Synthesis
I found this compound about 2 weeks ago wile reading through whimsy, it was in a thread labelled "List of compounds that are too much of a challenge".
Anyway i saw this as a challenge and after combing through the compounds listed there capsazepine stuck out like a saw thumb.
This stuff looks waaaaaaay to fun to not at the very least discuss the preparation for but please don't give me any shit like "why don't people post
practical procedures and do some real lab work instead of just coming up with theoretical preparations, i miss the old days when SM members spent more
time in the lab than in front of the computer".
Because this stuff is really hard to make and also as i will highlight extremely dangerous at some points so their aint no way in hell im gonna try
preparing it without first running through the practical implication with you guys first and even then im not sure if I am gonna make it any time soon
as much as id love too.
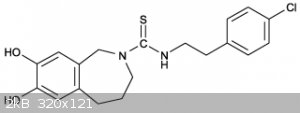
Anywhooo My idea was to start from pyrocatechol obviously followed by protection of the dihydroxy groups in order to carry out a grignard reaction, to
protect the dihydroxy groups i would propose either williamson with dihalomethane to prepare 1,3-benzodioxol or williamson with iodomethane to prepare
1,2-dimethoxybenzene followed by bromination with n-bromosuccinamide.
The reason i suggest benzodioxol over dimethoxybenzene is because from what i understand demethylation of vanillan is an absolute bitch and
demethylation of benzodioxol is as simple as refluxing in a strong acid such as HBr or HCl but id like to hear you guys thoughts on this?
Once the bromide is prepared and dried it would then be reacted with magnesium turnings in diethyl ether or THF to prepare a grignard reagent, this
grignard reagent would then be filtered and put into an addition funnel and dripped over 2-bromo nitroethane followed by acidic work up in the usual
fassion to prepare 3,4-methylendioxy-phenyl-2-nitroethane or 3,4-dihydroxy-phenyl-2-nitroethane.
Which ever was prepared would then need to be demethylated to reform the dihydroxybenzene followed by reduction of the nitro group using either
thiourea dioxide or sodium dithionite or catalyst/hydrogen, Id recommend thiourea dioxide simply because of ease of use and availability despite never
having used it myself.
The 2-bromo-nitroethane could be prepared by bromination of 2-nitroethanol which may again be prepared by the reaction of formaldehyde and
nitromethane.
https://de.wikipedia.org/wiki/2-Nitroethanol
http://www.orgsyn.org/demo.aspx?prep=CV5P0833
The next compound gives me pause as its a short chain thiol.
This compound is 3-hydroxy-1-propanethiol which sigma has named 3-Mercapto-1-propanol:
http://www.sigmaaldrich.com/catalog/product/aldrich/405736?l...
I unfortunetely have no idea as to how potentially dangerous this compound is to handle but i have an idea for its preparation. First reacting allyl
alcohol with HBr to get allyl bromide then reacting allyl bromide with sodium hydrosulfite to prepare allyl thiol which could then be reacted with HBr
in the presence of peroxide in a reverse markovnikov to get both 2-bomo-1-propanethiol and in higher yields the desired 3-bromo-1-propanethiol, Sigma
says the bp of 3-Chloro-1-propanethiol is 145*C and so the bromide should be a few tens of degrees higher, The 2 and 3 bromo propanthiols could be
hopefully separated via simple distillation, fractional might be necessary but i personally have never tried fractional distillation at those kind of
temperatures.
Sodiumhydrosulfite however can be prepared by reaction of hydrogen sulfide with sodium alkoxide, Scary.
http://www.sigmaaldrich.com/catalog/product/aldrich/c68601?l...
Once the 3-bromo-1-propanethiol and 3,4-dihydroxyphenyl-2-ethylamine is prepared neucliophilic alophatic substitution between both componants while
keeping the bromide at an excess in order to limit di and tri alkyation.
The unreacted amine and the di and tri alkylamine would then have to be separated although their shouldn't be to much to separate if done carefully
enough, since distillation is out of the question for these kinda compounds i suspect liquid layer chromatography will have to be employed 
The next part is where it get interesting.
https://www.heterocycles.jp/newlibrary/payments/form/00401/P...
This would employ a pummerer rearrangement to close the ring followed by reduction of the thiol using nickel chloride and sodium borohydride to close
the benzazepine and yield 5,6-dihydroxybenzazepine 
Unfortunately i think this may also yield 4,5-dihydroxybenzazepine but i don't have access to the paper.
Ok now this is where things get difficult, we need to substitute the amine with thioamide and i can only find 3 ways of doing this first is the
kindler thioamide synthesis which would have to probably follow N substitution with acetyl chloride.
http://www.organic-chemistry.org/namedreactions/willgerodt-k...
The next method used a rather difficult to prepare reagent called the lawesons reagent or possibly phosphorous pentasulfide, by reacting either of
these with the preprepared carbamide which im still a little iffy on how to prepare and also combined with the general un availability of lawsons
reagent im not really gonna go into this method.
The third looks most promissing but also the most dangerous, it involves N substitution using cyanogen bromide and then proceeding in a similar manner
as to how thiourea is prepared:
https://en.wikipedia.org/wiki/Thiourea
The LiAlHSH should not be too difficult to prepare provide LAH is on hand and cyanogen bromide can be conviently prepared by reaction of bromine with
sodium cyanide.
NaCN + Br2 ---> BrCN + NaBr
https://erowid.org/archive/rhodium/chemistry/eleusis/cnbr.ht...
This leave us finally with the preparation of p-chloro phenyl-2-ethylbromide,
My idea for this was again simple, starting from styrene followed by chlorination with ferric chloride under dry conditions followed by distillation.
Bp of p chlorostyrene is 192*C
http://www.chemspider.com/Chemical-Structure.13465.html
Bp of o chlorostyrene is 189*C
http://www.chemspider.com/Chemical-Structure.14205.html
For separation of the ortho and para isomers i recommend taking advantage of sterric hinderance in the same way that nerdrage did in his separation of
chlorotoluene video only at much higher temperatures, around 200*C maybe a bit higher in a dean stark set up, hopefully they don't decompose to much
at these temps.
The other much more lengthy method would be para nitration of styrene using acyl nitrate in zeolite:
http://www.sciencedirect.com/science/article/pii/S0167299199...
This p-nitrostyrene would then have to be reduced to an amine followed by diazatization with sodium nitrite and then finally reaction with copper
chloride to get p-chlorstyrene.
The p-chlorostyrene could then be reacted with HBr in the presence of a peroxide again in a reverse markovnikov to get a mixture of phenyl alpha and
beta brominated substrates, however im pretty sure phenyl-alpha-bromoethane undergoes hydrolysis to yield phenyl-alpha-hydroxyethane which would make
separation considerably easier, possibly simple distillation if we can find the bp of p-chlor-phenyl-alpha-hydroxyethane.
https://www.alfa.com/en/catalog/A16839/
Then to finally arrive at our final destination neucliophilic alophatic substitution of p-chloro-phenyl-beta-bromoethane with
N-thiourea-5,6-dihydroxybenzazepine wile keeping the benzazepine in excess to yield Capsazepine.

|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Would be nice (and probably would bring more resonance) if you would provide a scheme containing the whole synthesis route including arrows with
reagents, solvents, and reaction conditions. That would help greatly to understand your route and to identify potential problems faster..
Just as a small comment, I think LAH is a problem to get. It looks like most reagents in the synthesis need to be prepared first. Would be a huge
amount of work.
EDIT:
Another point is the Grignard reaction with 2-bromonitroethane will lead to attack of the nitro group, additionally the grignard reagent may simply
deprotonate 2-bromonitroethane leading to elimination of bromide with nitroethene as the product. Grignards usually are quite strong bases.
EDIT2:
2-Bromonitroethane will lead to the wrong chain length anyway, doesn't it?
[Edited on 6-5-2017 by Alice]
[Edited on 6-5-2017 by Alice]
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
I think you're making it much more complicated than it needs to be, and as Alice mentions you wouldn't even get the right product assuming it all
worked. In fact I gave up reading when I noticed this.
Anyway, what makes catechol the "obvious" starting material for this? To my trained eye, I'd suspect either vanillin or eugenol to be better
precursors, seeing how you're able to incorporate either the single or three sequential carbons of the azepine ring (respectively) and the "diol" is
already half protected. Both also provide functionality amenable to introduction of the nitrogen for the azepane ring also. They are probably easier
to aquire, too.
[Edited on 6-5-2017 by DJF90]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
The best way to make the compound would be through a convergent synthesis.Divide the molecular into 2 halves at the bond between the thiourea C and
the benzazepine N
Synthesis of the right half:-
1a) Convert fenclonine to p-chlorophenacetonitrile - https://www.sciencemadness.org/whisper/viewthread.php?tid=65...
2a) p-chlorophenacetontrile to p-chlorophenethylamine - http://pubs.acs.org/doi/abs/10.1021/ol403668e
3a) p-chlorophenethylamine to the isothiocyanate - http://www.tandfonline.com/doi/abs/10.1080/00304949209355899
Synthesis of the left half:-
1b)convert safrole to MD-butanoic acid (pg 5 of pdf) - https://www.jstage.jst.go.jp/article/cpb1958/44/3/44_3_500/_...
2b)intramolecular cyclisation of MD-butanoic acid to the ketone - http://www.tandfonline.com/doi/abs/10.1080/00304949109458304
3b)Modified schmidt reaction on the ketone - http://onlinelibrary.wiley.com/doi/10.1002/cmdc.201000101/fu...
4b)reduction of the lactam (pg 8 of pdf) - http://www.sciencedirect.com/science/article/pii/S0040402001...
If eugenol is used,an additional methylation step has to be done
1c) methylation of eugenol- http://www.sciencedirect.com/science/article/pii/S0040403998...
2c)O-methyleugenol to the butanoic acid - same ref as in 1b)
3c )intamolecular cyclisation of the acid to ketone - http://www.tandfonline.com/doi/full/10.1080/0039791080266339...
4c)modified schimdt - same ref as in 3b)
5c) reduction of the lactam - same ref as in 4b) or http://pubs.acs.org/doi/abs/10.1021/jm00039a006 (pg 2,scheme 1)
Putting it all together - react the right and the left halves -
http://onlinelibrary.wiley.com/doi/10.1002/chem.201102097/fu...
| Quote: | | Isothiocyanates are weak electrophiles. Akin to the reactions of carbon dioxide, nucleophiles attack at carbon |
https://en.wikipedia.org/wiki/Isothiocyanate#Synthesis_and_r...
here,the isothiocyanate is the right half and the nucleophile is the benzazepine N of the left half.
finally,deprotect the diol of safrole -http://pubs.acs.org/doi/abs/10.1021/jo9910740
eugenol demethylation (also works for safrole) - http://pubs.acs.org/doi/abs/10.1021/jo961191k
I have 2 doubts for the deprotection step:-
1. could HBr be used or would it also destroy the thiourea bridge ?
2. should the deprotection be done at the end or can it be done before putting together the 2 halves (i.e would the diols be nucleophilic enough to
compete with the bezazepine N to react with the isothiocyanate ? )
[Edited on 7-5-2017 by CuReUS]
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
| Quote: |
Would be nice (and probably would bring more resonance) if you would provide a scheme containing the whole synthesis route including arrows with
reagents, solvents, and reaction conditions. That would help greatly to understand your route and to identify potential problems faster..
|
If i can figure out a good place to host photos i can draw it all out and then post photos of that on here but i am having trouble finding a good way
to do this, starting to feel like an computer illiterate old person.
I unfortunately cannot find usable pictures online for most of the scheme either.
| Quote: |
Another point is the Grignard reaction with 2-bromonitroethane will lead to attack of the nitro group, additionally the grignard reagent may simply
deprotonate 2-bromonitroethane leading to elimination of bromide with nitroethene as the product. Grignards usually are quite strong bases.
|
I was afraid this might be the case i had found a few examples of nitros reacting with grignards but these were always aromatic and most organics
literature doesn't talk about the reactions of nitros with grignards.
As for the deprotonation, is the grignard able to deprotonate due to the close proximity of the electron withdrawing nitro, i know their are examples
of condensation between halo arenes and halo alkanes but im not sure about their limitations.
| Quote: |
2-Bromonitroethane will lead to the wrong chain length anyway, doesn't it?
|
Holy shit you're right, i just realized an enormous screw up on my part, although this does at least make a mannich reaction feasible for forming the
phenylmethylamine chain.
| Quote: |
Anyway, what makes catechol the "obvious" starting material for this? To my trained eye, I'd suspect either vanillin or eugenol to be better
precursors
|
I did look at vanillin briefly but due to my error in assuming the wrong chain length of the phenylalkylamine I had trouble figuring out how to
prepare the phenethylamine from the benzaldehyde. The other issue i had with vanillin was that it was methylated and i didn't like the idea of
demethylating the methoxide to hydroxide although in hindsight the pyridine/thiourea and AlCl3 are probably some of the easiest things to acquire in
this synthesis.
This makes thing a hell of a lot easier, as you can see by my origional idea starting from styrene there where way to many issues with high
temperatures, I like the idea of forming the thiourea bridge from isothiocyanate as this nullifies the danger of pissing around with dangerous
thiolating reagents. The only pain in the ass part i see here is gettin your hands on carbon disulfide but if i recall correctly their are a few
rather OTC ways to make carbon disulfide floating around here on the forum.
You don't specify as to how you would form the benzazepine ring from the phenylpropylamine so im assuming this is specified in the reference you gave,
i unfortunately don't have access  but i will put a request in References. but i will put a request in References.
Thanks for all your replies.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Alice this might help
I was having issues uploading the photos from my camera, for some reason they registered as being to big but i have managed do this using my phone so
apologies for the bad quality, also I don't have much light so they arn't the easiest to read.
This is my origional idea however i haven't drawed out the route to the p-chloro-phenyl-2-bromoethane.
I skipped drawing out the reduction of the nitro group and the deprotection/demethylation of the diol.
I will try drawing out the route suggested by CuReUS soon.
Again given recent posts this method wont work, however it may work if we were to replace the grignard between the bromobenzene and
2-bromo-nitroethane with possibly a mannich reaction between catechol ammonia and formaldehyde however im not sure if this would be specific enough to
just alkalate the desired position and we may end up with 2,3-dihydroxy-phenylmethylamine as a side product.
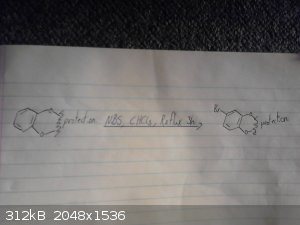 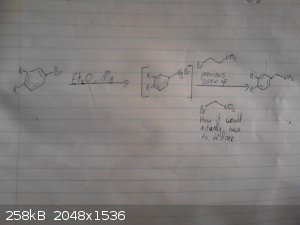 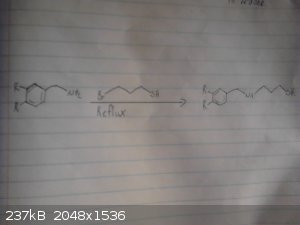 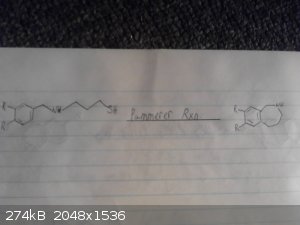  
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Assured Fish, thanks for the pics. Now as you can see, in the second pic, there is a carbon vanishing.
For drawing such schemes, either use the chemdraw trial version or the ChemSketch freeware.
For the bromination, NBS is not OTC, but as this is an activated arene there is a vast number of halogeation reactions available. Br2 being one of
them or just in situ generated Br2.
| Quote: | | As for the deprotonation, is the grignard able to deprotonate due to the close proximity of the electron withdrawing nitro, i know their are examples
of condensation between halo arenes and halo alkanes but im not sure about their limitations. |
Yes, either there is a concerted elimination of HBr or the negative charge is resonance-stabilized by the nitro group before elimination happens. See
Henry reaction where deprotonation is intended in order to generate a nucleophile. Bromonitromethane won't solve the problem either as it's
deprotonated too.
For the next step the reagent you suggest is 3-bromopropane-1-thiol. This will also react with itself and reacts twice with the benzylamine, as such a
reaction would be performed with an auxiliary base, otherwise HBr formed blocks benzylamine for further reaction or may deprotect the phenolic groups.
A short search reveals for such reagents the thiol is often protected as thioacetate.
For the other routes, CS2 isn't the only problem. For the other reactions stuff like phosgene, diborane, TiCl4, ethyl chloroformate, NaN3, POCl3,
trichloroacetic anhydride, and TEMPO are required. Reagents difficult to make or pretty much dangerous to handle. This is hardly pratical and for
being OTC something more straight forward and easier would be nice.
Additionally, the most crucial part for all of these reactions isn't the reactions themselfes or the various reagent preparations, but how to purify
the products and how to make sure the compounds are the compounds intended. I count about 10-11 transformations not including the reagent
preparations.
[Edited on 7-5-2017 by Alice]
|
|
|
UserPrevKnownAsVanta
Harmless
Posts: 10
Registered: 18-4-2017
Member Is Offline
Mood: No Mood
|
|
Chemdraw pls
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Not directly,but it can be made from OTC chemicals -http://www.sciencemadness.org/talk/viewthread.php?tid=3366
succinic acid is sold as food and dietary supplement - https://en.wikipedia.org/wiki/Succinic_acid#Food_and_dietary...
| Quote: | | For the other routes, CS2 isn't the only problem. For the other reactions stuff like phosgene, diborane, TiCl4, ethyl chloroformate, NaN3, POCl3,
trichloroacetic anhydride, and TEMPO are required. Reagents difficult to make or pretty much dangerous to handle. |
In case you haven't noticed,I have changed my initial route.You only need NaN3 and TiCl4 now.Azide is OTC or can be made from
OTC chemicals - http://chemistry.mdma.ch/hiveboard/acquisition/000435713.htm....As for the latter,it can be bought.And you don't neccesarily have to use
TCAA,using that just gives the best yield.Other cyclising agents could also be used.
| Quote: | | This is hardly practical and for being OTC something more straight forward and easier would be nice. |
stop suffering from OTC OCD. .This isn't a nitrobenzene to aniline type of
synthesis.Desperate compounds call for desperate measures. .This isn't a nitrobenzene to aniline type of
synthesis.Desperate compounds call for desperate measures. | Quote: | | Additionally, the most crucial part for all of these reactions isn't the reactions themselfes or the various reagent preparations, but how to purify
the products and how to make sure the compounds are the compounds intended. I count about 10-11 transformations not including the reagent
preparations. |
instead of laughing at other people's syntheses,why don't you come up with a "simple, straight forward,practical and easy route" which uses safe
reagents and is 100% OTC ?
[Edited on 7-5-2017 by CuReUS]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
CuReUS, I just thought that something has changed. BTW I'm not laughing at other people's synthesis at all. That was just my way expressing this
synthesis isn't trivial - not bad. 
Of course, with enough energy and time it's possible achieving a lot OTC, but if really nothing is available and everthing has to be prepared in one
or multiple steps in a more or less complicated manner it's just getting impossible for normal people to afford the workload.
Fortunately I have indeed made my own thoughts on the problems I have named.
Here is my draft for the synthesis of the left part (benzazepine). I paid much attention making the synthesis OTC friendly using only chemicals which
are either available or there are established OTC procedures for making them. I'm aware the last step (thermal cyclization) is the most critical step
and may be substituted in one way or another, maybe by cleaving the benzyl ether beforehand. The preparation of 1,3-diiodopropane will take two steps.
1. Transformation of 1,3-propanediol by distillation from NaCl and a suitable acid into the dichloride. 2. Double Finkelstein reaction with NaI
leading to Cl/I exchange.
EDIT: Forgot to provide the link for yeast mediated reduction of vanillin:
http://pubs.acs.org/doi/abs/10.1021/ed070pA155
EDIT3: Added more precise wording.
EDIT4: Update
In chemistry there is always room for optimization, a never ending story.
Possible side products of 1,3-dichloropropane (b.p. 121 °C) synthesis like allyl chloride (b.p. 45 °C) or 1,2-dichloropropane (b.p. 95 °C) will be
removable by fractionated distillation, alcohols by washing the product multiple times with water. I assume removing the chlorinated products under
reduced pressure during the reaction may be fortunate or even neccessary. I have chosen the chlorination over iodination or bromination because the
boiling points are more practical and it's better starting with cheap stuff.
The Finkelstein reaction may be performed with just 1 eq. NaI, as the statistical yield of 3-iodo-1-chloropropane is 75%. Assuming a slight dependence
on the second substituent, e.g. sterically, an even higher ratio may be obtained. This leads to a lower boiling product which might then be distilled
under reduced pressure (aspirator vacuum) saving half of the sodium iodide. The Grignard will prefer substituting the iodide over the chloride while
the chloride remains still reactive towards ammonia. The ammonia additon may be done one-pot, or just after removal of the salts, as it enables
further acid-base extraction. At that point hopefully other side products not being an amine will be removed. A possible toxic side product is
propylene-1,3-diamine which is water soluble and removed by acid base extraction.
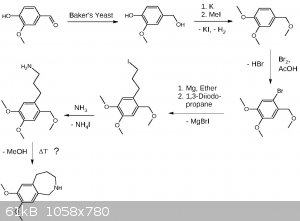
[Edited on 7-5-2017 by Alice]
[Edited on 7-5-2017 by Alice]
[Edited on 8-5-2017 by Alice]
[Edited on 8-5-2017 by Alice]
|
|
|
Eddygp
National Hazard
Posts: 858
Registered: 31-3-2012
Location: University of York, UK
Member Is Offline
Mood: Organometallic
|
|
I doubt you would lose methoxy like that in the 7-m. cyclisation step. Maybe a sulfonate ester would be better. In any case, a friend of mine actually
devised a very plausible route to capsazepine, I'll redirect him to here.
there may be bugs in gfind
[ˌɛdidʒiˈpiː] IPA pronunciation for my Username |
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@Eddygp. As I said I didn't suggest the last step because it sounds so promising, it would just be so interesting to know whether it works. As a
temperature range I think about anything between 200 °C and pyrolysis. At least for me chemistry would become very boring if I never tried to check
out where the limits are.
On the other hand the benzyl ether can be transformed into any other functional group, for example back to the aldehyde, which would give an imine
then. Still easier to reduce than the lactam though. A quaternary benzylamine would be another idea.
[Edited on 8-5-2017 by Alice]
|
|
|
12thealchemist
Hazard to Others
Posts: 181
Registered: 1-1-2014
Location: The Isle of Albion
Member Is Offline
Mood: Rare and Earthy
|
|
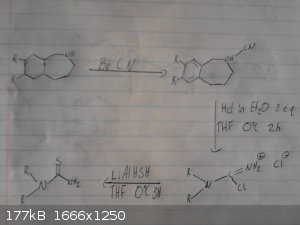
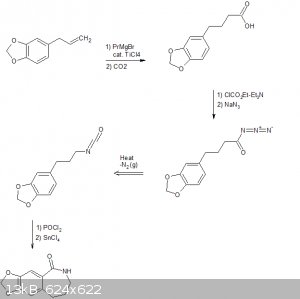
As per request, I'll attach my synthesis here, starting from vanillin and styrene.
There are two possibilities for the reduction of the oxime because I found two different methods on YouTube by competent chemists. In writing a more
detailed document, I included both of these.
Also, the use of boron tribromide as the demethylation agent was chosen since there was an OrgSyn reference for that procedure, and both boron and
bromine are relatively OTC.
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
12thealchemist, I have four comments on your synthesis pointing out potental problems.
1. Styrene will yield mostly markovnikov product upon treatment with HCl.
2. For the alkylation of the benzylamine the question is if this stops at the secondary amine.
3. For the Friedel-Crafts mediated cyclization I suspect the intramolecular cyclization being less favored compared to intermolecular alkylation of
another molecule's ortho-phenol position which might or might not be overcome by highly diluted solution. A phenol is more activated than a
methoxybenzene.
4. Not sure if BBr3 is capable of chopping off the alkyl from the 7-membered ring again leading to some undefined tar but I don't know how the
reaction conditions mayt be, favoring one reaction over the other. The Friedel-Crafts alkylation is reversible in general, leading to difficulties
which limit its synthetic use. But I haven't checked the literature about how the scope of this individual reagent BBr3 is.
Finally the synthesis of the benzylamine and the isothiocyanate looks very good!
|
|
|
12thealchemist
Hazard to Others
Posts: 181
Registered: 1-1-2014
Location: The Isle of Albion
Member Is Offline
Mood: Rare and Earthy
|
|
Alice:
1. I was aware that styrene + HCl would give mainly the undesired product; I had forgotten to point that out in the post. My thinking was that since
the precursors were cheap and readily available, one could obtain sufficient quantities for the main synthesis without too much difficulty. However, I
appreciate the drawbacks of this idea, namely green chem.
2. Crossed-fingers
3. Not something I had given much thought to; the lecturers at my university have historically indicated that intramolecular reactions are kinetically
favoured over intermolecular ones. However, your point raises another one - could it instead react ortho to the phenol instead of meta? This does form
a bridged compound, however.
4. The orgsyn reference:
Org. Synth. 1969, 49, 50
DOI: 10.15227/orgsyn.049.0050
http://www.orgsyn.org/demo.aspx?prep=CV5P0412
[Edited on 8-5-2017 by 12thealchemist]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Or unfortunately,as we will soon see
First of all,any route is only as good as its references.Since you do not provide suitable references,your route fails there itself.
Now lets go through your synthesis
1.For the vanilin reduction using yeast,not everyone has a centrifuge lying around,you know.
2.You have problem using azide,but no problem using K ?Also MeI is a very safe reagent according to you ?
3.bromination will give other isomers too.Will you be able to separate them ? Also,the HBr formed as a side product might demethylate the ethers
4.Pls give a reference where an alkylation is performed using grignard alone
5.How will you prevent the formation of 2nd,3rd and quaternary amines ?
6.And last but not the least,The cyclisation you propose might work in wonderland,but it won't work here on earth
Quote: Originally posted by Alice  | | On the other hand the benzyl ether can be transformed into any other functional group, for example back to the aldehyde, which would give an imine
then. Still easier to reduce than the lactam though. |
What do you mean by "still easier to reduce".Do you mean the process or the reagent ? Because if its the former,the amount of work you would have to
do to convert the benzyl ether to the amine(selective de-alkylation,controlled oxidation,imine formation,reduction while trying to prevent reductive
alkylation of the amine by the aldehyde) will outweigh the effort required to reduce the lactam.And if its the latter,and you are shy of using LAH,you
could just use Red-Al instead..
| Quote: | | A quaternary benzylamine would be another idea. |
What do you plan to do with that ?
Your route is quite simple,but the problem is that in organic chemistry,simplicity and selectivity do not go hand in hand
| Quote: | | There are two possibilities for the reduction of the oxime because I found two different methods on YouTube by competent chemists.
|
I don't know which competent chemists you are referring to,but both the chemists I know have run into problems trying to reduce oximes using
Zn.Chemplayer said that the final product was inpure and had to be furthur purified whereas nile red failed to reduce it itself.Another youtuber
claims to have done it,but he got a shitty yield of 36%
2.In the alkylation step,isn't there a chance of the propane dichloride reacting twice with NH2 to form an azetidine ?
3.Even if the FC intramolecular cyclisation went well,that naked phenol and even the NH would react with the AlCl3 to form tar
4.Demethylating the ether before reacting it with the isothiocyanate might lead to a competing reaction between the OH and the N,as I have mentioned
before
As for the other half,you will have a warm time during the p-chlorination.Just ask Nurdrage.Also the last step is wrong,since isothiocyanates are
extremely susceptible to hydrolysis,and the NaOH would react with it before the benzazepine even had a chance
[Edited on 9-5-2017 by CuReUS]
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
If you lack drawing software: https://anonym.to/http://py-chemist.com/
====================================
edit: reading is hard
From safrole, treatment with Cp2ZrHCl followed by benzylamine in THF results in anti-Markovnikov hydroamination to
5-(3-benzylaminopropyl)benzodioxole.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3800136/
This reacts with formaldehyde in aqueous acid in a Mannich reaction to give N-benzylbenzodioxolo[5,6-c]azepane.
N-benzylbenzodioxolo[5,6-c]azepane is then debenzylated with Pd/C/H2 or Pd/C/triethylammonium formate.
This is treated with p-chlorophenethyl isothiocyanate to yield capsazepine methylenide. The methylenedioxy group can be cleaved by a number of
standard methods such as:
https://www.jstage.jst.go.jp/article/cpb1958/42/3/42_3_500/_...
Safrole itself can be made by a number of methods; generally the bromination of benzodioxole:
BzdH + NH4Br + H2O2 [HOAc] >> BzdBr
https://erowid.org/archive/rhodium/chemistry/aromatic.bromin...
followed by the formation of an organozinc reagent in acetonitrile and reaction of this with allyl acetate:
BzdBr + Zn + CoBr2 [MeCn] >> BzdZnBr
BzdZnBr + allyl-OAc >> safrole + "ZnBrOAc"
http://pubs.acs.org/doi/abs/10.1021/ol0340641
would seem preferable based on what people don't want to be ordering from chemical companies (piperonal, eugenol, IBX, etc).
If safrole is an issue it may be possible to substitute 2,2-dimethylbenzodioxole made IIRC by the condensation of catechol with acetone. However I
know less about the cleavage of this moiety. 2,2-dimethylsafrole is not illegal AFAIK (it is not a drug precursor)
[Edited on 9-5-2017 by clearly_not_atara]
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
Hi CuReUS.
Not getting my point isn't a reason behaving like a child as you have illustrated:
| Quote: | Or unfortunately as we will soon see
First of all,any route is only as good as its references.Since you do not provide suitable references,your route fails there itself.
Now lets go through your synthesis |
This also includes use of majestic plural and not setting white spaces properly in order to not provide good readability. If you won't stop this I
will ignore you until you're acting like a grown up again.
I thought you're the one not being happy with laughing about other people's synthesis. I didn't, but you seem to have some weird double standards.
A route fails only if it doesn't work in the end. Reactions not working as expected may be modified or substituted. Only if that fails the route
fails. There is nothing special about this. My route is as hypothetical as yours, no matter if it's based on literature procedures or not. And now for
the third time, I'm aware the cyclization as shown in my draft is experimental!
| Quote: | | 1.For the vanilin reduction using yeast,not everyone has a centrifuge lying around,you know. |
Surely a centrifuge is the only way extracting vanillyl alcohol. :/
| Quote: | | 2.You have problem using azide,but no problem using K ?Also the MeI is a very safe reagent according to you ? |
I never said I have a problem working with azides, MeI or K. In fact I worked with all of them multiple times. I said it's a huge amount of work
together with a lot of other comparably difficult procedures making it hardly affordable for many. Therefor the procedure for making MeI is a lot
easier than making azide. But it's true MeI in particular doesn't suit the toxicity issue I have raised. I won't work with MeI at home as I don't
have a fume hood, but others have a fume hood so handling MeI is relatively safe as long as the person handling it is trained and ventilation works
properly. My reasoning to suggest MeI is simple preparation and effectiveness.
| Quote: | | 3.The bromination will give other isomers also.Will you be able to separate them ? |
I don't know many aromatic substitutions which don't give mixtures so I would be surprised if this reaction wouldn't give a mixture too. Fortunately
substituents already there are leading to a more or less good regioselectivity, additionally the reactivity of the electrophile affects
regioselectivity too. Making the benzyl ether in order to enhance regioselectivity isn't a coincidence. If you have a look at the substrate, there is
only one possibility for a para-methoxy substitution having additionally the benzyl ether ortho which makes it the most activated position. Therefor
the product of interest is formed in excess. Did you realize I was applying the rules of regioselectivity prediction? Further, recrystallization is
the method of choice purifying the product.
| Quote: | | 4.Pls give a reference where an alkylation is performed using grignard alone |
I don't need a reference for my own hypothesis. My aim is starting as simple as possible, and only if this fails choosing a more advanced/less
amateur-friendly alternative. So I'm familiar with the various additives and alternatives, I just want to use them only if it's really neccessary.
This way I'm trying to end up keeping the route easy and practical.
| Quote: | | 5.How will you prevent the formation of 2nd,3rd and quaternary amines ? |
By using an excess of hot conc. ethanolic ammonia, with the halide being droped slowly into it. This means choosing a suitable amount of ammonia to
outperform multiple alkylations of the product.
| Quote: | | 6.And last but not the least,The cyclisation you propose might work in wonderland,but it won't work here on earth |
I don't share your attitude of knowing beforehand what's possible and what's not. Once I heard a lecture of a professor who said to the students: if
you are done with the master, try to forget as much as possible as it will just clog up your mind.
Of course that was meant polemic but the core message is: never be too sure.
| Quote: | | What do you mean by "still easier to reduce".Do you mean the process or the reagent ? Because if its the former,the amount of work you would have to
convert the benzyl ether to aldehyde(selective de-alkylation followed by controlled oxidation) will outweigh the effort required to reduce the
lactam.And if its the latter,and you are shy of using LAH,you could just use Red-Al instead.. |
No, the latter, making LAH or whatever needed. LAH or Red-Al aren't OTC and even if you have any source it still doesn't make it OTC in general. Maybe
OTC has a different meaning for each of us. That may explain a lot.
| Quote: | Quote:
>> A quaternary benzylamine would be another idea.
What do you plan to do with that ? |
As an alternative starting material for thermal cyclization. Trimethylamine may be a better leaving group than methoxide.
| Quote: | >> Quote: Originally posted by 12thealchemist
>> As per request, I'll attach my synthesis here, starting from vanillin and styrene.
Your route is quite simple,but the problem is that in organic chemistry,simplicity and selectivity do not go hand in hand |
This wasn't directed to me, but I answer anyway: The search for simplicity even for complicated problems is one of the aims of green chemistry.
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: | | 4.Demethylating the ether before reacting it with the isothiocyanate might lead to a competing reaction between the OH and the N,as I have mentioned
before |
The isothiocyanate will not react with the phenol, as phenols do not autoionize in non-aqueous solvents, so it is okay to deprotect before this
coupling. I think a couple of people missed this. As another example, ammonium acetate is ionized in water but neutral in DMSO. The amine-isocyanate
coupling would almost certainly be performed in a less polar solvent like methylene chloride.
======================================================================================================
Alice -- some thoughts
Grignard reagents will not generally react with alkyl halides without a copper catalyst.
I'm confused why you've chosen a likely problematic thermolysis (such reactions usually require a Lewis acid and occur in the gas phase, also often
water is the product and the amine is inserted into the ether) over simply protecting the aldehyde with ethyene glycol and using intramolecular
reductive amination (generally an easy reaction) at the end. You need a (likely acidic) deprotection step anyway after all. This also avoids using any
alkali metal.
| Quote: | | excess of hot conc. ethanolic ammonia |
I suggest you plug in the Henry's law constant calculated in this paper, estimate that the partial pressure of ammonia won't rise above half an
atmosphere (and you better not have any sparks anywhere...), and calculate the corresponding solubility. I'm not expecting encouraging numbers.
http://www.sciencedirect.com/science/article/pii/S0378381211...
The Delepine reaction is more feasible.
Generally your route is constructed with a very lax attitude to flammability, which is strange for someone who claims to have experience working in a
home lab. I would not recommend an air-free technique (such as the Grignard or potassium) to anyone who can't prepare cuprous iodide. By contrast I
even recommended an alternative for hydrogen. It's something you might want to consider. Many home chemists die in fires.
[Edited on 9-5-2017 by clearly_not_atara]
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Why not first read about what has already been accomplished rather than pissing in the wind about a whole lot of chemistry that ain't going to work.
Lithiated β-aminoalkyl sulfones as mono and dinucleophiles in the preparation of nitrogen heterocycles: Application to the synthesis of capsazepine
DA Alonso, A Costa, B Mancheño, C Nájera - Tetrahedron, 1997 - Elsevier
The lithiation of N-benzyl-β-tosylethanamine (10a) and N-benzyl-α-phenyl-β-
tosylethanamine (10b) with n-butyllithium at− 78° C leads to monoanions 11a and 11b, respectively. Intermediates 11 react with different
monoelectrophiles (D2O, alkyl halides,
Cited by 31 Related articles All 4 versions Cite Save
A Facile and Practical Synthesis of Capsazepine, a Vanilloid Receptor Antagonist
J Lee, J Lee - Synthetic communications, 1999 - Taylor & Francis
Page 1. SYNTHETIC COMMUNICATIONS, 29(23), 4127-4140 (1999) A FACILE AND PRACTICAL SYNTHESIS OF CAPSAZEPINE, A VANlLLOlD RECEPTOR ANTAGONIST Jeewoo
Lee* and Jiyoun Lee ... ABSTRACT: A facile and practical synthesis of capsazepine, a vanilloid receptor
Cited by 9 Related articles All 3 versions Cite Save
An efficient parallel synthesis of capsazepine and capsazepine analogs
L Tafesse, DJ Kyle - Combinatorial chemistry & high throughput …, 2004 - ingentaconnect.com
Capsazepine (CPZ, 1) is a well-known vanilloid receptor (VR1) antagonist that has been
cited widely used inthe literature. However the current synthetic methods used for the total
synthesis of CPZ are lengthy, involve multiple purification steps, and produce low yields.
Cited by 7 Related articles All 4 versions Cite Save
Maybe then you can speculate on alternate routes.
AvB
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Aaaaaand im late to the party.
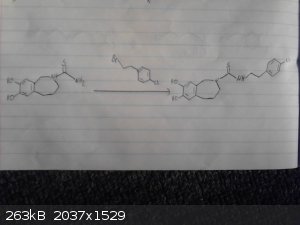
The following scheme is what I think was origionally proposed by CuReUS starting from safrole or eugenol, however i am a little confused as to how you
intended to use a modified schmidt reaction on the ketone/amide, i thought we would just want to reduce this completely using NaBH4 or LAH to get the
benzazepine ring.
 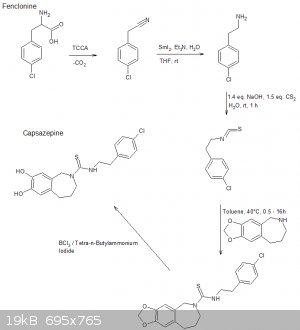
The formation of the benzazepine ring was taken from this reference that was sent to me from CuReUS. http://pubs.acs.org/doi/abs/10.1021/jm00039a006
Fenclonine to nitrile. http://www.sciencemadness.org/talk/viewthread.php?tid=32534
And his reference for the deprotection of the diol for which i don't have access to. http://pubs.acs.org/doi/abs/10.1021/jo9910740
I have a modification though for which i have not drawn out unfortunately.
Instead of forming the isothiocyanate from the amine using CS2 we could instead opt to convert the amine to an isonitrile using chloroform and a base
which would be about as OTC as you get:
CHCl3 + NaOH/KOH -----> CCl2 + R-NH2 -----> R-NC
The isonitrile could then be reacted with sulfur like in the following reference:
http://www.organic-chemistry.org/abstracts/lit4/754.shtm
This would mean that we could do away with the CS2 and instead opt for an OTC approach using sulfur, chloroform and KOH to bridge the
p-chlorophenethylamine and benzazepine forming the thiourea bridge.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
Some references
AvBaeyer i managed to get your second reference but i cannot find your first one and ive yet to get access to the third: lee1999
CuReUS this is your demethylation reference: brooks1999
Attachment: lee1999.pdf (371kB)
This file has been downloaded 554 times
Attachment: brooks1999.pdf (156kB)
This file has been downloaded 938 times
|
|
|
Alice
Hazard to Others
Posts: 111
Registered: 11-5-2015
Member Is Offline
Mood: No Mood
|
|
@atara, I know about the Grignard issue. But my stance is to check out if this particular reaction will work this way. In my previous post I explained
why I think this is a good idea.
| Quote: | | I'm confused why you've chosen a likely problematic thermolysis (such reactions usually require a Lewis acid and occur in the gas phase, also often
water is the product and the amine is inserted into the ether) over simply protecting the aldehyde with ethyene glycol and using intramolecular
reductive amination (generally an easy reaction) at the end. You need a (likely acidic) deprotection step anyway after all. This also avoids using any
alkali metal. |
I never said what I've drawn in the draft is the most promising possibility. After all discussion can make a synthesis just becoming better, not
worse. Acetal protection may be a very good idea and very OTC. The question is what influence it will have on bromination and cross-coupling. The
question has to be answered for the benzyl ether too of course.
| Quote: | | Generally your route is constructed with a very lax attitude to flammability, which is strange for someone who claims to have experience working in a
home lab. |
Not sure what you mean. I don't think I've ever stated having much of a home lab. I don't think a ~10 step synthesis is suitable by any means for non
trained experimenters. Where trained includes refusing doing potentially dangerous steps without proper equipement and safety measures. It also
includes knowing when to grab the fire extinguisher and when to run. For working with ether, especially diethyl ether, a sufficiently strong airflow
is advisable. But as a side note, diethyl ether introduces the fortune being good in moderating temperature as it cools quite strong upon evaporation.
Additionally a Grignard doesn't have to be neccessarily done in an inert atmosphere as the ether protects the liquid more or less from air contact.
Nevertheless inert gas is the better choice if available.
| Quote: | I suggest you plug in the Henry's law constant calculated in this paper, estimate that the partial pressure of ammonia won't rise above half an
atmosphere (and you better not have any sparks anywhere...), and calculate the corresponding solubility. I'm not expecting encouraging numbers.
http://www.sciencedirect.com/science/article/pii/S0378381211...
The Delepine reaction is more feasible. |
It would be highly advisable collecting ammonia in some washing bottles anyway. You're right about the solubilitity. Doesn't sound very encouraging,
but solubility isn't neccessarily a prerequisite. Bubbling ammonia through the solution might still do the job, especially for the iodoalkyl.
Sure, Delepine would provide a higher chance of success without much experimenting. Thanks for mentioning.
| Quote: | | I would not recommend an air-free technique (such as the Grignard or potassium) to anyone who can't prepare cuprous iodide. By contrast I even
recommended an alternative for hydrogen. It's something you might want to consider. Many home chemists die in fires. |
I wouldn't recommend any organic multi-step synthesis to someone not being able preparing cuprous iodide.
[Edited on 9-5-2017 by Alice]
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
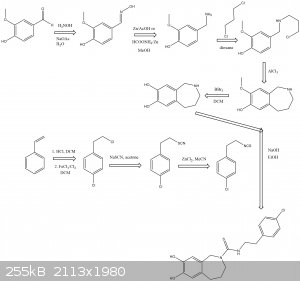
Possible ways for synthesis Capsazepine by reaxys
Attachment: Capsazepine Synthesis.pdf (106kB)
This file has been downloaded 493 times
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
learn to accept criticism.You go around poking holes in other people's syntheses but get offended when others do the same to you.What fun!
| Quote: | | This also includes use of majestic plural and not setting white spaces properly in order to not provide good readability. |
I used plural because I wanted to point out the flaws in your route to others.As for the spaces,blame the site posting software,not me
| Quote: | | My route is as hypothetical as yours, no matter if it's based on literature procedures or not. |
My route is logical and backed up by refs,whereas yours uses basic reactions hoping as if by miracle that they will give react exactly the way you
want them to.
| Quote: | | Surely a centrifuge is the only way extracting vanillyl alcohol. :/ |
that's what they use in the only ref you linked,possibly to remove the crappy yeast cells.Don't shoot the messenger
| Quote: | | Did you realize I was applying the rules of regioselectivity prediction? |
No I didn't,I got into chemistry yesterday..Also since the ring in highly
activated,you will have di/tri bromination to care of too
| Quote: | | Further, recrystallization is the method of choice purifying the product. |
that's easier said than done for some isomers,if you don't have the proper refs detailing the separation.Just ask nurdrage https://www.youtube.com/watch?v=_wbqqI0IgY8&index=9&...
| Quote: | | I don't need a reference for my own hypothesis. My aim is starting as simple as possible, and only if this fails choosing a more advanced/less
amateur-friendly alternative. So I'm familiar with the various additives and alternatives, I just want to use them only if it's really neccessary.
This way I'm trying to end up keeping the route easy and practical. |
so basically you want to do trial and error instead of saving time and money following well established research.An easy and practical route is
useless if it doesn't work.
| Quote: | | By using an excess of hot conc. ethanolic ammonia, with the halide being droped slowly into it. This means choosing a suitable amount of ammonia to
outperform multiple alkylations of the product. |
I don't think you know how much NH3 you would need to prevent polyalkylation if you decided to follow that primitive route.IIRC,the ratio
of NH3 to RX is 20:1 .Have fun working with that much
NH3
| Quote: Originally posted by clearly_not_atara | | The isothiocyanate will not react with the phenol, as phenols do not autoionize in non-aqueous solvents, so it is okay to deprotect before this
coupling. I think a couple of people missed this. |
actually,I had a doubt about this upthread.But since no one clarified it,I assumed it to be a potential side reaction,to be on the safer side.I am
relieved that the deprotection can be done before the final coupling.It would have been a disaster to destroy the whole molecule just to take the
protecting group out
| Quote: | | The amine-isocyanate coupling would almost certainly be performed in a less polar solvent like methylene chloride. |
They used acetonitrile in the paper I linked - http://onlinelibrary.wiley.com/doi/10.1002/chem.201102097/fu...
Man,your compound seriously heated up this thread
| Quote: | | The following scheme is what I think was origionally proposed by CuReUS starting from safrole or eugenol, however i am a little confused as to how you
intended to use a modified schmidt reaction on the ketone/amide, i thought we would just want to reduce this completely using NaBH4 or LAH to get the
benzazepine ring. |
Brilliant drawing,but you have messed up a few steps
1.In the 2nd step,the acid undergoes an intramolecular FC acylation using TCAA to form the ketone
2.Then the modified schimdt is done on the ketone using HCl/NaN3 to get the cyclic amide(lactam)
3.Finally,the lactam is reduced to benzazepine using LAH
As for the right half-
1.I just realized now that there is no need to convert fenclonine to the nitrile and then reduce it,it can be directly decarboxylated to the amine
https://erowid.org/archive/rhodium/chemistry/tryptophan.html
http://www.sciencemadness.org/talk/viewthread.php?tid=8574
2.The safrole is deprotected before the coupling,following atara's recommendation.
3. As I mentioned above,acetonitrile is used as the solvent in the coupling step,not toulene. Did you have any reason for using the latter ?
| Quote: | | Instead of forming the isothiocyanate from the amine using CS2 we could instead opt to convert the amine to an isonitrile using chloroform and a base
which would be about as OTC as you get |
from the ease with which you suggest making isonitrile,its obvious that you have never worked with them.I suggest you don't,they stink to high heavens
and they are extremely toxic(I got poisoned from just one whiff the first time I made them)
[Edited on 9-5-2017 by CuReUS]
|
|
|
| Pages:
1
2
3 |
|









