isothiazolinone
Harmless

Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
Need help recovering alpha-phenylacetoacetonitrile
So, thru the help of many of the chemists on these forums, I have managed to synthesize a fairly good amount of phenylacetonitrile. From that, via
sodium ethoxide and anhydrous ethyl acetate i have successfully made the sodium salt that comes from this. I literally spent hours drying it on the
vacuum pump, did not have diethyl ether....well I did...but I opted to use dcm because it is much easier to produce obviously and much safer in my
estimation cancer considerations aside. So, I get to the point where I have this moist sodium salt, that has been washed with dcm, so I add water to
it to get it into solution, place it in a freezing mixture made of water, salt, ice, acetone and methanol, get the temperature down to around -5 C.
Ok, so I go to add the glacial acetic acid.....add it in and nothing.....its almost as if the sodium salt has disappeared. I know the salt is in there
because I added it to water an hour prior, but adding the glacial acetic acid does not seem to precipitate it. I thought maybe its not cold enough do
I threw it back in the freezing mixture to see if anything would fall out but nothing. I have tried adding straight glacial acetic acid as well as
diluted glacial acetic acid. Kinda miffed here. Does anyone have any advice.
|
|
|
isothiazolinone
Harmless
Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
whoops...sorry for my initial missteps in forum ettiquette (part deux)
Whoops...in my thirst for knowledge I may have neglected proper forum etiquette. I initially posted this topic before but got absolutely 0 response,
and im realizing now it may be in large part to the fact that I did not post any references to illustrate the methodology i was following and quite
possibly appeared to be seeking this information for free without doing any preliminary work. Either that or this topic was pre-destined to be cast
into deutritus in which case im sure i just sealed the fate of this topic by double posting this and ill stop wasting everyone's time. My apologies in
advance if my ACS source citing is not perfect, must have overslept for those classes in college.
Followed this initially.
]https://erowid.org/archive/rhodium/chemistry/phenylacetone.html
First synthesis down on the web page.
Went about the drying of the ethanol slightly different though instead of calcium oxide followed by distillation with sodium metal, I opted for
initial dehydration with anhydrous magnesium sulfate followed by treatment with 3a molecular sieves. I dried the ethyl acetate in the manner
suggested.
That synthesis, as far as I can tell, is pulled from the much more robust and informational article here.
Julian, Percy L, Oliver, John J, Kimball, R.H., Pike, Arthur B, Jefferson, George D. A-Phenylacetoacetonitrile. Org Synth 1938, 18, 66 http://www.orgsyn.org/demo.aspx?prep=CV2P0487
So after following the path of the two synthesis above, I was able to successfully get to the point where I had a significantly large portion of the
sodium salt separated and isolated on the buchner funnel. So now the synthesis calls for saturating the salt with water, washing the water with dcm or
ether (i used dcm) I am assuming this is to remove any phenylacetonitrile that did not react, and then neutralizing the solution of the base and
enolate by adding glacial acetic acid to allow the B-Keto ester to form. The problem is, that when i do this, nothing happens. Its almost as if all
the sodium salt that i just added to the water and hour prior has basically disappeared. ( i should add that after saturating the sodium salt with
water, i placed the flask in a salt water freezing bath in my freezer for an hour). Fail!.
There is also modification of the synthesis here...
https://erowid.org/archive/rhodium/chemistry/p2p.phenylaceto...
I should note I followed this as well and this is the only path I had success with. Albeit limited success The primary difference with this route
being that you skip the initial salt separation via vacuum filtration and instead opt for direct saturation of the sodium salt with water after it is
allowed to sit for a period of time. From what I understand, a key factor in a successful claisen condensation is letting the reaction mixture rest
for a certain period of time to allow for completion of the reaction although I can't remember why. When I followed this pathway, when I added the
glacial acetic acid there was an instant and noticable milky white precipitate that formed which I was able to separate and isolate on the funnel. The
problem was is that my final yield was somewhere in the neighborhood of 21 grams, down to 15 grams after recrystallization with methanol. I did only
perform this reaction at 1/2 the suggested starting materials but even so the yield is a far cry from what is projected.
I even went so far as to seek out this article, that is pre-1900 that seems a bit antiquated in its methodology, but nonetheless seemed to have some
relevant info in it. Its in German, so I used Google Translate for the english translation, which was still helppful, although its quite funny how all
of the really important scientific vernacular remains in German...might need to brush up on my German chemistry language skills which at present are
non-existent...i mean im no dimwit..i can put 2 and 2 together and figure out what alpha-phenylacetessigester means but all the same. Anyways, that
article is here.
Ueber den alpha-Phenylacetessigester
Walter Beckh
Chem. Ber. 31, 3160-3164 (1898)
Other than what I found in these, there seems to be very limited info on the synthesis of this compound specifically, although I did find out quite a
bit of info about claisen condensations. One follow up question i had too concerns the neutralization of the enolate and the base to form the B-keto
ester as the final step. I noted that as sort of a general guide line for this type of reaction it suggests the use of either sulfuric or phosphoric
acid for the neutralization step, where as in three of the four articles listed above it suggests the use of glacial acetic acid....which is what I
used. Is there anything to be gained from using sulfuric or phosphoric acid, besides (im assuming) the ability to most likely need less sulfuric or
phosphoric acid than gaa. Also, is it possible to in effect over neutralize the solution and turn it too acidic to make it hard or impossible for the
B-keto ester to form?
I should qualify all this by saying that i am by no means a highly educated chemistry student, I did take organic chem in college...but long story
short my current job is installing home irrigation drip systems...if thats any indication of how the whole organic chem thing went. Most of my
chemistry knowledge has been self taught in the last few years, largely by stalking this forum for information. I find forums to be the most helpful
and useful because they seem to cut out all the bullshit and sort of reduce the dividing line between whats realistic in a fully stocked well funded
laboratory and whats practical for a home hobbyist chemist. Anyways, sorry for my initial misstep in forum etiquette. I am not just trying to seek
this information for free, I think I have done my due diligence in terms of preliminary research, but i have hit a roadblock and have not been able to
find the help or answers I seek elsewhere. If anyone who knows more about this than me would be willing to lend a helpful hand it would be much
appreciated. Thanks in advance for your help....or for tearing me to shreds and casting me into deutritus....either way.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
You got ignored basically because it's about making a methamphetamine precursor.
We're kind of a not-for-profit entity, and drugs (of that nature) are unequivocally for profit.
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
This isn't the most common approach. But, this seems like a legitimate question. And heck, you might even be in a jurisdiction where such
manipulations are legal.
So, here's the tip.....Follow the Experimental Details, EXACTLY as presented. Exactly!
Often, actual yields are crap, even then!
Now, as to acid...... In general, Acetic Acid is in no way a substitute for Sulfuric Acid, or Phosphoric Acid. Acetic Acid is a weak acid, and
Sulfuric Acid is about as strong as you can get. Though I would'nt contradict an Organic Synthesis Procedure.
Others here, would be more adept at explaining the actual technical mechanics involved. And, maybe they will. If you ask the right questions.
Not a procedure I personally have performed. Check Via the search engine here. Or, peruse the actual Hive Archive, which has recently been
resuscitated. Might be lengthy discussions recorded there.
https://the-hive.archive.erowid.org/
[Edited on 5-11-2016 by zed]
[Edited on 5-11-2016 by zed]
|
|
|
isothiazolinone
Harmless
Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
Thanks for answering the question. Obviously i dont have any sway or pull here as obviously i dont have very many posts or time on this forum. I
certainly don't want to step on any toes, but if im not mistaken i don't believe i asked a question a/b converting this ester to benzyl methyl ketone.
I certainly understand that an interrogative pathway to that compound would certainly be undoubtedly indicative of my intentions. I realize that, and
thus I purposely chose to omit that line of questioning...of course thats assuming that i have any interest to pursue that compound to begin with. Im
not the first person on these forums to stumble upon a hard to acquire compound from time to time. As I stated prior, I am a hobbyist chemist and not
a very skilled one at that. I know this about myself, and for that reason i chose to pursue the conversion of phenylacetonitrile to the ester because
i was not too fond of the idea of trying to hydrolyze phenylacetonitrile to say phenylacetic acid.....another kind of no doubter this is my intention
precursor, a reaction....that if im not mistaken also produces cyanide gas....which going back to my lack of skill....I was not too fond of being
anywhere in the vicinity of. I preferred to take my chances with sodium ethoxide instead, cause last time I checked you can survive a lungful of that
compound. I have successfully completed multiple grignard reactions in the past (hint. I find that while keeping everything as dry as possible is
certainly integral to the success of that reaction, i have found from personal experience that whats more important is pretreating the magnesium with
elemental iodine on a hotplate stirrer at around 100 c under a steady and inert stream of nitrogen or some other inert gas, and using a large stir bar
to intermittantly mar and crack any surface oxidation built up on the magnesium to expose fresh metal. This simultaneously avoids the need to add an
iodine crystal to catalyze the reaction later, and also basically ensures that the grignard kicks off instantaneously avoiding the lengthy times it
takes for the reaction to start that so many experience.) My point in explaining my experience with the grignard is that to get that experience I have
done it many many times. I knew that there was a reaction using the grignard mechanism to convert phenylacetonitrile to benzyl methyl ketone, but
since I had done so many grignards in the past, and because i have no interest in the compound that comes from that, I refrained from pursuing that
route with the phenylacetonitrile because i wanted something new to try. Im sure there are other routes and cool things to do with phenylacetonitrile,
this is merely the route I chose to explore and i would just like to see the time I invested come to sort of successful fruition. Any judgment beyond
what i just said is nothing more than pointless, speculative pontification and in my estimation a complete waste of my time and the other members who
have been kind enough to address my concerns free from judgement (see Zed above)  . .
Zed, first off let me say that I have read many many of your posts at length in the past and have always found them to be very helpful and
informative. Its actually kind of funny that you were the one that answered this post because i also live in the state of Jefferson, albeit about 4
1/2 hours south down I-5. ok i see what you have said above, and I have a few follow up questions for based off that I am hoping you would be kind
enough to help me with. So, when you said that this isnt the most common approach to this compound, to me that means that there is a much more obvious
or approachable route to follow....is this something that i may have missed? I saw that there was a kind of alternate approach to a compound similar
to the alpha-nitrile on orgsyn here http://www.orgsyn.org/demo.aspx?prep=CV2P0284, but i noted that it seemed you had to already have the alpha-nitrile in hand to perform the
synthesis...which obviously thats the compound im trying to get too currently. Also, when you tell me to follow the experimental details exactly, is
that something you know to be very specific to this reaction mechanism, or are you telling me this in more of a "follow the experimental details to
the exact presentation before freewheeling it" kind of way. Either reason is valid to me just curious. Now, another follow up question in regards to
what you said about the acid. The orgsyn approach I mentioned above does in fact use glacial acetic acid as the acid in lieu of either phosphoric or
sulfuric acid. I only knew about the sulfuric acid/ phosphoric acid possibility from sort of a generalized outline I read on performing claisen
condensations, but that was non-specific towards any one compound. The german patent i mentioned above I believe also chose sulfuric acid. You stated
that glacial acetic acid is weaker than either phosphoric or sulfuric acid, which I certainly have found to be true and agree with. What I find to be
strange though is it seems that the chemists from the references above knew glacial acetic acid to be weaker, and decided to use it for that very
reason, but the only times I have had any form of success with this reaction (although as i stated very limited success), was when I added undiluted
glacial acetic acid to complete the formation of the B-Keto ester. Don't necessarily know what that means, but it seems relevant and is certainly a
noteworthy observation i have made in the past. You said you don't have any personal experience with this reaction, but it does certainly seem like
you know more about it than i do so I had just one more question. Its about the reasoning for making the solution as cold as possible. Is the point of
that more about about aiding the formation of the ester or is it more in an attempt to make the compound and/or any remaining ethoxide less volatile.
The reason i ask is because when I was able to get the ester to successfully form, the solution was at a temperature no where near freezing, and was
at more of say somewhere halfway between freezing and the average ambient temperature indoors in the southern state of jefferson in the middle of
september. call it approximately 5 degrees C. Anyways, sorry for rattling on at length about this, but you have extended the one and only life line I
have had on this synthesis so im really trying to grab it. Any more light you could shed on this or any specific links you think would be helpful
would be much appreciated. I have poured thru the hive archives as well as the archives of this forum as well as the others that deal with similar
topics such as these and haven't been able to turn up anything else but perhaps i will go back and try to dig a little bit deeper. Thanks for your
help by the way.
|
|
|
Texium
|
Threads Merged
6-11-2016 at 07:46 |
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Iso,
Hard to read. Many words....Few paragraphs. Is it possible to use fewer words, and more paragraphs?
To begin with, to my way of thinking......the hydrolysis of Phenylacetonitrile to Phenylacetic Acid, should not release HCN gas. It should proceed to
the Amide, and then to the free Acid.
http://www.orgsyn.org/demo.aspx?prep=CV1P0436
As to creating your current material. No, I don't really know an alternate route.
And No, experimentally, I wouldn't freelance at all. When performing an unfamiliar synthesis; follow the experimental details EXACTLY. If they say
to use KOH; use KOH.....not NaOH. No substitutions. If the procedure calls for K, use K.....not Na. Zero C means Zero C.. Negative 30C, means
Negative 30C.. Don't try to wing it.... Go out and buy some dry ice.
If the experimenters use Ethyl Acetate as a solvent, but you don't have any on hand.......don't substitute! Go out and get some Ethyl Acetate.
Still no guarantee that your procedures will work properly, but your odds are improved.
As for legality...proceed at your own peril. Some of the posters here, do what appear to be wild things. But, the manipulations they perform are
legal in their individual jurisdictions. Others are licensed, or are experimenting in an academic or industrial environment, where their activities
are legally permitted.
Me? I like being a free man, and I don't need money. I personally, wouldn't engage in "Breaking Bad" type behavior. Prison sentences these days,
are in decades, not years.
[Edited on 6-11-2016 by zed]
[Edited on 6-11-2016 by zed]
[Edited on 7-11-2016 by zed]
[Edited on 7-11-2016 by zed]
|
|
|
isothiazolinone
Harmless
Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
Sorry, im still not up to snuff on the proper etiquette of this forum, in the future I will be sure to break up my posts so they are easier to read.
Once i start rattling on i just become too concerned with getting everything out i need to say, at the cost of not making it very easy for the people
trying to help me read what it is im saying.. my apologies
I also agree with what you say in terms of the acid hydrolysis of the nitrile to phenylacetic acid, like I said i have no desire to toy with that at
all because i have less than zero desire to produce the compound that comes from that reaction, i was merely mentioning that because i was giving
examples of reasons why i was chosing not pursue other syntheses with the nitrile.
In terms of what you said of following the experimental details exactly, I suppose that is the one thing i have not tried yet. I have either gone
about the drying of the ethanol with magnesium sulfate and molecular sieves instead of the suggested route with calcium oxide and subsequent sodium
treatment. Which actually brings me to a question about some confusion I had using sodium metal to dehydrate ethanol. I have made sodium ethoxide in
the past which involves dissolution of sodium metal in ethanol. I guess my question is...if i am supposed to use sodium metal to dehydrate the
ethanol.....how am i just drying the ethanol but not making sodium ethoxide? Is it more of a molarity thing where im keeping a very high molar excess
of ethanol to sodium metal to promote alchohol dehydration and limit ethoxide formation. Or am i basically resigning myself to the idea of making
sodium ethoxide which also dehydrates the ethanol at which point im just basically distilling away the newly formed anhydrous ethanol from the
ethoxide?
Aside from all that, i will follow your suggestion and follow the experimental details to the absolute T and dotted I, hopefully that will be the
missing link so to speak. Additionally, on a hunch last night and with the thought that the ester formation may be more contingent on a more neutral
ph than I thought, I added sodium carbonate to the solution to bring it very close to neutral while still being acidic and then put it into the
freezing mixture to see if i could precipitate it. I will report back to share the results of that to let anyone interested know if that was
worthwhile or not.
Finally, I appreciate your wise words and concerns over the legality of what I am doing. As I stated prior I dont have any desires to to take this
compound any further than the alpha-nitrile but all the same still very much so appreciate your advice. Thanks again. Will be sure to report back.
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Regarding the Ethanol and Sodium. Now, this all seems like extra work to me too. But, I yield to the procedure of the authors. They knew what they
were doing.
Relatively dry Ethanol may be made anhydrous by refluxing it with Sodium, and distilling it directly into the flask wherein it is going to be reacted.
This flask should be protected by drying tubes. Hard to keep Ethanol really anhydrous, if you are going to allow it to be exposed to ordinary "wet"
air.
So, you are leaving behind any NaOH and Sodium Ethoxide, formed during refluxing...And, you are starting fresh with Anhydrous Ethanol, which you then
react with fresh Na to create "Clean" Sodium Ethoxide. Seems redundant, but the authors seem to have done it that way. And reports indicate, that
the procedure "Works" when it is performed in that manner.
To my way of thinking, a solution containing Dry Ethanol and NaOH cannot be Completely Anhydrous. There is an equilibrium present wherein EtOH+NaOH
> EtONa + H2O
Since this reaction cannot tolerate H2O. It would appear that NaOH should also not be present.
I'm kinda tired and confused right now, and this isn't really my field.... So I reserve the right to completely recant.
P.S. It is a day later. Going back further, I note that you did not use ether, as recommended by the authors. But instead, made a substitution.
Generally speaking, Organic Synthesis procedures are the golden standard. They work really well.......but only, if you follow them to the letter.
This you have not done. Try doing so, and get back to us with your results.
[Edited on 9-11-2016 by zed]
|
|
|
isothiazolinone
Harmless
Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
Thanks so much for your patience zed and more importantly thanks for the time you have taken to respond to my queries. I really appreciate it more
than i can tell you. While this may not be your field, i still feel like you know considerably more than i do in terms of the generalities of hows
things work and more about the guts behind why these reaction mechanisms work. Admittedly one of my major scholarly flaws is that I am not always
neccessarily a linear learner which tends to be how the entire western world is standardized in terms of its educational values. Sort of like a
pyramid where there is a solid foundation and then levels are built upon that foundation and where one level of the pyramid is contingent upon the one
below it and structurally can't exist without the level below it. Im more of a "necessity is the brother of invention" type learners where i sort of
jump ahead to what i need to do out of necessity and then back track my way to the beginning learning all the fundamentals and mechanisms after the
fact. Not the best way to do things and i think its clear that this has been detrimental to my successful synthesis in this reaction.
My plan is to follow the experimental details exactly on this next attempt. So regardless of my opinions i already am in process of dehydrating the
ethanol with calcium oxide....to do that I have heated the calcium oxide in my oven at the highest temp it could muster for around 56 hours, and then
I added that to the 190 proof ethanol im using and am going to leave the calcium oxide in solution with the ethanol for 24 hours. My plan then is to
separate out as much of the lime as i can or i guess what would be calcium hydroxide at that point on the buchner, and then add the sodium to the
ethanol, reflux and then perform a simple distillation with the receiving/ future reaction flask protected by a gas bubbler with concentrated sulfuric
acid in it to keep out moisture. The part of the experiment i actually did follow precisely was the dehydration of the ethyl acetate by treating it
with phosphorus pentoxide for an hour and then refluxing it for 1/2 an hour and then distilling it ...so im going to follow that pathway again....as
it was set out in orgsyn. I suppose we shall see what I can get this time following the details closely.
P.s. in terms of the ether vs dcm usage. I opted for the dcm for many many reasons. First, the article that is linked from the rhodium post on this
compound which is claimed to be higher yielding used dcm instead of ether. Second, dcm is much more easily obtained than diethyl ether. And i mean
this not just in terms of purchasing, but I also mean in terms of diy methods of obtaining it. Ive never felt the need to do a distillation of paint
stripper to obtain dcm in my shower to help mitigate the damage of an explosion or fire, cant say the same for distilling ether from engine starter
fluid or from synthesizing ether from ethanol and sulfuric acid. Plus, i have read it and heard it said countless times that just about everyone who
has enough experience with diethyl ether has been victim too or witness too some sort of mishap with it whether they work in a lab or not. I count
myself included in that statement, although i was quite lucky as only a small amount of fumes had turned to peroxides in a small flask so after the
flash flame there was no flask full of liquid ether to ignite as well and I was able to get the flask reasonably far away from myself before the
explosion and avoided any injury...did have protection on however. Anyways, my point with that story is I already had more than healthy fear of ether
leading up to that occurrence and took all the necessary precautions and still almost paid dearly
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
TL; DR
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Iso, the more I read about this reaction, the less I understand.
There seem to be few references on this reaction, and not much available regarding the actual reaction mechanism itself. Personally, I like to see
things mapped out, and I haven't been satisfied with what I have found so far.
As for Hive posts.....Do keep in mind, that Hive posts are sometimes fictions.
And Ether, yeah, I try to avoid it too. But, I have never had an incident with it. When I'm using Ether, I make damned sure there are no flame or
spark sources, anywhere even remotely nearby. Light switches? I tape them over, so they can't be flipped.
Peroxides? In my day, reagents were cheap, and I could just roll over to the nearest chemical supplier, and buy myself a fresh can of Ether.
You are doing better, but pay heed to stoichiometric-steve's bottom note:
TL;DR.... Apparently, this abbreviation predates text-speak. It is an editors note..... It means, Too Long;Didn't Read.
Odds are, there is someone here that knows something about this reaction. But, your posts have been long and complicated. Hard to read.
[Edited on 9-11-2016 by zed]
|
|
|
Mesa
Hazard to Others
Posts: 264
Registered: 2-7-2013
Member Is Offline
Mood: No Mood
|
|
Fairly sure this has been the most popular/common route for the last 10 years or so(discounting commercial mexican/dutch superlabs)... It's
consistently been the (2nd) most common chemical I see chinese suppliers on alibaba etc. advertising. I wouldn't be surprised if it's scheduled here.
|
|
|
kmno4
International Hazard
Posts: 1496
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
I performed this synthesis in a few runs, with many modifications.
Originally it was given in OS in 1938 and it is a little outdated.
It means that you can use many alcohols (methanol, ethanol, isopropanol, butanol....) and esters (ethyl acetate, buthyl acetate.....). Conditions may
not be strictly anhydrous, because traces of water (from starting components, mainly from solvent) are consumed at once in hydrolysis of applied
ester.
Nevermind. Prepared sodium salt (most possibly solvated) of the ketonitrile* is soluble in water, but quickly hydrolyses and solution becomes turbid
during several minutes (at room temp.)
When any acid is added, ketonitrile is precipitating immediately.
I used acetic acid, HCl, H2SO4, H3PO4 (all diluted with water) with the same effects.
If you have no precipitate..... it means that something is totally wrong. Are you sure of you cyanide and EtOH ?
I used phenylacetonitrile prepared by myself, distilled at normat pressure (yes!) without significant decomposition/polymerization.
It has rather pleasant smell, but during long-term storage it darkens, so it is not perfectly pure.
* drying of this salt is loosing your precious time, when it is washed and filtered as much as possible, no dryinf is needed for hydrolysis step. DCM
is good, dioxane is good, toluene is good.....
Слава Україні !
Героям слава !
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
No offense, Iso, but you'd probably get better results if you'd just drop the whole "I have no intention of making *insert drug/precursor here*,
honest!" act. We've all seen and heard it a thousand times before. It'd also help if you'd refrain from going off on random tangents about unrelated
things and posting enormous walls of text that look suspiciously similar to an amphetamine-induced rant. Now, if you really are just
innocently trying to make alpha-phenylacetoacetonitrile for shits and giggles, then I do apologize; however, we aren't idiots, and you aren't the
first newcomer to appear out of nowhere and immediately express interest in some obscure precursor that has very little real application outside of
illegal drug synthesis.
And just so it's clear, we don't actually care what you intend to do with the alpha-phenylacetoacetonitrile, so there's really no need to keep
reassuring us that you have no intention of making phenylacetone. Generally speaking, as long as your intentions aren't motivated by profit or
hurting/maiming/killing others, you'll find that most here are more than willing to help you synthesize whatever. Just keep the discussion scientific
and chemistry-oriented and do as much research beforehand as possible and you'll more than likely get the help you need.
So anyway, what exactly is it that you're trying to do? Do you want the nitrile, or do you want its hydrolysis product, alpha-phenylacetoacetic acid?
You keep mentioning "beta-keto ester," but alpha-phenylacetoacetonitrile is not an ester. Yes, the reaction between the ethyl acetate and
phenylacetonitrile is essentially a Claisen condensation; however, the product in this case is not a beta-keto ester because the condensation is
between an ester and a nitrile, not two esters. Thus alpha-phenylacetoacetonitrile is instead a beta-keto nitrile. A beta-keto
ester, on the other hand, would be something like ethyl alpha-phenylacetoacetate.
Quote: Originally posted by zed  | Iso, the more I read about this reaction, the less I understand.
There seem to be few references on this reaction, and not much available regarding the actual reaction mechanism itself. Personally, I like to see
things mapped out, and I haven't been satisfied with what I have found so far. |
I'm guessing there haven't been any real studies on the actual mechanisms at play. But if it helps, I've drawn a plausible mechanism for the
production of alpha-phenylacetoacetonitrile from ethyl acetate and phenylacetonitrile, as well as its hydrolysis to alpha-phenylacetoacetic acid and
subsequent decarboxylation to phenylacetone.
Ethyl acetate + phenylacetonitrile ==> alpha-phenylacetoacetonitrile:
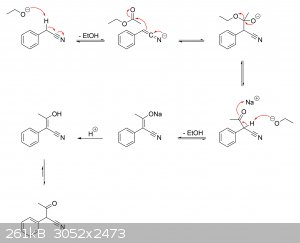
Alpha-phenylacetoacetonitrile ==> alpha-phenylacetoacetic acid ==> phenylacetone:
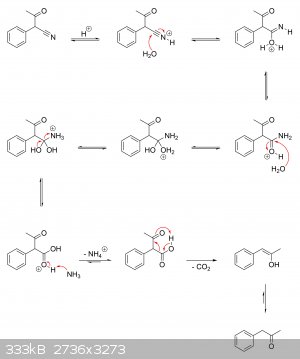
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Thank you Darkstar.
Mesa, Benzyl Cyanide listed? You bet! List 1.
It has been considered a questionable purchase, in the U.S., for about 50 years now.
[Edited on 10-11-2016 by zed]
[Edited on 10-11-2016 by zed]
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
This is my least favourite thread in all the forums. Not only does the OP completely fail to make himself clear, but ignores advice as to his chemical
endeavours as well as to the composition of his posts.
I suggest this is moved to detritus, especially with all the cookery involved.
|
|
|
isothiazolinone
Harmless
Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
My apologies to all. Someone offered me a helping hand and i grabbed it...and apparently ripped the arm off as well. The only other thing ill say
about non-chemistry related
topics is that as can be seen from this thread was the first response I received was an attack not a helping hand so i felt the need to defend myself.
While these forums are
great and i love them, an unfortunate by product is that we only have the ability to judge people based off of what they are asking for help with and
little else. And while I
could continue to rattle on at length in my own defense, I have been advised not too, plus i feel any other words i could say in that respect are not
likely to sway the
preconceived notions of people one way or the other and are thus not only a waste of your time but a waste of mine.
On topic, hopefully i can do this in a concise and proper enough way to not get my head ripped off.
-Zed, yes I have found things to be much the same as you...there seems to be very very limited information about this specific compound. There is
quite a wealth of
knowledge about the Claisen condensation but that is mostly broad generalities not dealing specifically with this synthesis. This lack of info is what
prompted me to come
here for help.
-kno4. Thank you, this is the type of info i really needed from someone with practical experience of synthesis. Rather than ask all the questions here
im hoping its ok that i pm you since i seem to be wearing on the patience of other members in this thread. That being said, if Im able to bring this
synthesis to a good end I will post the experimental details i followed to success in the hopes of helping someone else or perhaps getting more
insights on how it can be improved.
-to everyone else I apologize for my haste and missteps in forum etiquette i am not only a novitiate in chemistry but also a novitiate in this forum
and I have not quite learned a way to navigate the forums quite yet without testing the patience of the people trying to help me. Just like things can
be learned and improved upon in chemistry, i suppose I need to improve and learn how to succeed in these forums without pissing everyone off.
My apologies and my thanks.
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
stoichiometric-steve,
Iso's writing style needs improvement. Otherwise, there is nothing wrong with this posting. The OP is asking for assistance in performing a
synthesis, for which there are very few readily available references.
He doesn't seem to be a potential terrorist, as many of our newbie posters have appeared to be. And, he has been polite and decidedly non-abusive.
I, for one, have found this interaction very informative. And, I welcome Iso's future postings.
Iso, please keep us posted on your progress.
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
I concur; I don't think there's much wrong with this discussion.
If phenylacetone is what you're after, and you have benzyl cyanide (aka alpha-phenylacetonitrile), then you could just react it with a methyl Grignard
reagent (or methyllithium). You'll get the ketone after acidic workup. This would also work with acetonitrile and a benzyl Grignard.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
If the OP could clarify what his target molecule is then further assistance could possibly be provided.
|
|
|
kmno4
International Hazard
Posts: 1496
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
I decided to repeat my earlier experiments, beacuse I forget some experimental details and results.
Also because I found some forgotten benzyl cyanide in a bottle. It smels good, a little yellow but I think it is still OK for the reactions.
First reaction I repeated was: ethanol/etyl acetate/sodium/the nitrile.
Amazing: no condensation occurs 
Reaction mixture is white suspension of sodium ethylate, does not changing on heating. I do not know what is going on, but earlier it worked without
any problems. However, previously ethanol and the nitrile were freshy prepared/distilled.
Another run:
Instead of ethanol, isopropanol(IPA) was used: this goes normally. Reaction mixture becomes reddish colored soon as ester+nitrile was added to
sunpension of alcoholate. During refluxing, white solid is formed (as it sould be).
Another run:
IPA (~21 cm3),buthyl acetate (7,5 g), dioxane(~6 cm3), 5 g of the nitrile and 1,3 g of Na. Without dioxane, reaction mixture is hard to stirr
magnetically, white solid (sodium salt of ketonitrile) is often adhered to the bottom of RBF. Instead of dioxane, toluene, heptane,THF.... may be
used. It also increases yield of reaction (at least in theory).
Unfortunately, amount of prepared ketonitrile (in both cases) was rather low, below 40-50% of theory. Seems that a lot of cyanide is present in
post-reaction mixture.
As I remember, the yield was always above 60-70% in earlier runs. Wet reagents ? I am worried about total failure of ethanol run, because it means
(most possibly) that used alcohol is very wet. I will try to prepare some fresh sample of anh. ethanol and repeat the run.
But first I am going to find real water content in my ethanol, possibly by method presented in "A Simple and Fast Procedure for in situ Determination
of Water in Ethanol Fuel"(DOI: 10.5935/0103-5053.20130054)
Слава Україні !
Героям слава !
|
|
|
kmno4
International Hazard
Posts: 1496
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
Another, another run:
Mixure of IPA (5 g) and toluene (~21 cm3) was prepared and 1,3 g of sodium was dissolved* in this mixture.
Solution of butyl acetate (7,5 g) and 5 g of the nitrile was added to the hot suspension of sodium isopropanolate in toluene.
The reaction looks very promissing, white powdery sodium salt is separating at once from the reddish solution.
To complete the reaction, it was heated to weak boiling (+ mag. stirring) during 2-3 hours.
Sodium salt was filtered and washed 2x3 cm3 of toluene. Even at this moment it looks that amount of sodium salt is larger comparing to earlier runs.
Toluene wet salt weights 13,2 g, dried 8,0 g. It was then suspended in water, citric acid added to weakly acidic pH, filtered, washed, dried and
weighted.
Very nice yield of ketonitrile was obtained: 5,9 g with m.p. 90 C. It corresponds to about 90% of theory. This clearly shows that the original
procedure (and many other procedures from OS) is obsolete and can be improved by simple means.
* dissolving Na in such small amout of isopropanol is very slow, even on boiling. Small amount of Na may be still present during addition of
ester/nitrile mixture, but it is not harmful.
[Edited on 25-11-2016 by kmno4]
Слава Україні !
Героям слава !
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
So, did you decide to use the Sodium salt of Isopropyl Alcohol, instead of Sodium Ethylate.......In order to avoid the lengthy procedures, required
to produce truly "Dry" Ethanol? Well, if the reaction worked...Bravo! Good idea!
Also noted, that the change in solvent and reactive ester, would seemingly increase the reflux temperature applied to the reactants. Perhaps,
assisting the reaction towards completion....and thus producing an increased yield?
Ummmm. Methinks you should forward your results to OS. Looks like an interesting approach.
PS...Thank you!
[Edited on 26-11-2016 by zed]
|
|
|
isothiazolinone
Harmless
Posts: 11
Registered: 12-7-2016
Member Is Offline
Mood: No Mood
|
|
wow,thanks so much Kmno4. That amount of work and expertise is very helpful. I wanted to report success with this reaction as well. Currently working
on writeup of my experimental details and i will post it here shortly for peer review.
|
|
|
careysub
International Hazard
Posts: 1339
Registered: 4-8-2014
Location: Coastal Sage Scrub Biome
Member Is Offline
Mood: Lowest quantum state
|
|
| Quote: Originally posted by zed |
...
So, here's the tip.....Follow the Experimental Details, EXACTLY as presented. Exactly!
...
|
Interesting side note, this is what constituted the transformation of alchemy into chemistry - adopting the practice of writing precise technical
descriptions of procedures with all observations made of the process and products such that they were reproducible.
Georg Agricola did this with metallurgical processes in De Re Metallica in 1556, and Johann Rudolf Glauber with more general chemistry starting around
1625.
About that which we cannot speak, we must remain silent.
-Wittgenstein
Some things can never be spoken
Some things cannot be pronounced
That word does not exist in any language
It will never be uttered by a human mouth
- The Talking Heads
|
|
|
|








