tshirtdr1
Harmless

Posts: 31
Registered: 12-12-2014
Member Is Offline
Mood: No Mood
|
|
Synthesis of non-volatile esters
Hello all,
I am trying to synthesize ethyl esters of amino acids. These esters exhibit a higher boiling point than the solvent (ethanol). I know I can do this
with acid chlorides, etc. Additionally, I can just order them, but I am attempting to do this in a cost effective manner since I basically already
have everything that I need. I have attempted this synthesis a couple of times, but I lost it all in the workup.
I have attempted ethyl glycinate using dry HCl (bubbled through) and dry ethanol over and under sieves and nitrogen with refluxing for 5 hours.
I seem to get a good yield, but I tried distillation (BP = about 160C) and scorched it. I tried extraction, but I can't find a good solvent system
because if I neutralize the acid with sodium bicarbonate, the neutral ester is basic and I seem to be getting a reaction with acetone and ethyl
acetate (probably an aldol-type of reaction). I have not tried ether, as I do not have any fresh ether or THF. Also, I am afraid an aqueous workup
will hydrolyze the ester. I would be able to order ether, THF of MTBE if advised.
I would welcome suggestions. Please keep in mind that I am fairly new to real synthesis as my background is primarily computational, but I have basic
lab equipment and chemicals available.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
I don't understand what your problem with the workup is. I mean, it is obvious you didn't actually do anything proper to isolate the ethyl glycinate
hydrochloride, but what I don't understand is why didn't you follow the procedure from whatever reference you used.
| Quote: | | I seem to get a good yield, but I tried distillation (BP = about 160C) and scorched it. |
Amine hydrochorides are generally not volatile and thus they cannot be distilled.
| Quote: | | I would welcome suggestions. |
What's wrong with rotavaping and recrystallizing the hydrochloride?
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
tshirtdr1
Harmless
Posts: 31
Registered: 12-12-2014
Member Is Offline
Mood: No Mood
|
|
Thanks
Obviously, the problem is that I have no reference for this. I have seen that it can be done, but no real synthetic references. I am assuming that if
I simply rotovap, I will have a lot of impurities, especially some unreacted glycine. I have considered doing that. I guess I can try that next and
see how impure it is.
Thanks.
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
If you drive the reaction towards completion with lots of ethanol and several bubblings of HCl, then it should be nearly all product. At that time,
just bubble some N2 through the reaction to drive off most of the HCl, and then concentrate the reaction to a dry, white solid, which will be the
ethyl ester HCl salt. This can be used as is in the next step, in most cases, and you would simply add an equivalent of a non-reactive amine to
neutralize the HCl salt in the next step. Triethylamine is most often used, but many others might work. I have made many of these esters the same
way. You can often generate the needed HCl in-situ by adding a ml or so of acetyl chloride to the ethanol before adding the aminoacid.
Triturating the product with ether might help remove any minor impurities, but is not often needed. Recrystalizing the product would best be done by
finding it in a prep, CRC handbook, etc and seeing what others have already done to purify it, as it is a common chemical and rediscovering an ideal
crystallization condition is a lot of work.
Note: You would pretty much never use acetone in an organic prep workup, especially if doing any sort of aqueous work up, as it is miscible in water
and will make a mess.
|
|
|
tshirtdr1
Harmless
Posts: 31
Registered: 12-12-2014
Member Is Offline
Mood: No Mood
|
|
Thanks Dr. Bob
I will try your suggestions, especially bubbling N2. Thanks. I didn't think of that.
I only used acetone D6 for NMR, and I noticed a reaction, but not for extraction. I just didn't add that detail.
Thanks again.
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Quote: Originally posted by Dr.Bob  | You can often generate the needed HCl in-situ by adding a ml or so of acetyl chloride to the ethanol before adding the aminoacid.
|
Yes; I've done this myself with glycine and methanol in one of my classes. It works great, but in an amateur situation acetyl chloride is generally
too much of a pain to get to justify using it to make HCl. Instead, you can make anhydrous HCl from salt and sulfuric acid.
Edit: It was phenylalanine, not glycine. Here's the procedure:
Acetyl chloride (1.5 eq, 1 vol.) is added slowly to ice-cooled MeOH (6 vol) over 20 min. L-phenylalanine (1 g) is added, and the reaction mixture is
stirred overnight. On the next day, remove the solvent with a rotating evaporator to produce a white solid, which is then dissolved in water. Add
slowly solid NaHCO3 until the solution is not acidic.
(The white solid after rotavapping is the hydrochloride salt; adding the bicarb is necessary for the next step of the synthesis, which is reaction
with ethyl chloroformate to form a carbamate.)
An aqueous workup does not hydrolyze the ester (at least not for phenylalanine), and it's worth a try.
[Edited on 4-7-2016 by Metacelsus]
|
|
|
clearly_not_atara
International Hazard
Posts: 2800
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Obviously OP has a way of making HCl or he wouldn't already be using it.
You might be able to ensure complete (or very nearly complete) reaction by drying the reaction solution over MgSO4 part-way through the reaction
(since the reaction produces water), and then bubbling HCl one more time and waiting another hour or so. I doubt it will be necessary but it's worth a
try if you have issues crystallizing the ester post-reaction.
[Edited on 6-4-2016 by clearly_not_atara]
|
|
|
tshirtdr1
Harmless
Posts: 31
Registered: 12-12-2014
Member Is Offline
Mood: No Mood
|
|
Thanks a lot!
I appreciate everyone's assistance. I am not in an amateur situation, but I am, sadly, an amateur at synthesis. My PhD is in computational chemistry,
but I teach at a small college and am doing this for a small research project involving undergraduate students. I have limited funds and my equipment
is not very advanced, so I try to stay away from dangerous chemicals. I do, however, have a glove box, a nitrogen line, and spectroscopy equipment to
play with.
I think my main problems were that I was doing this with sieves in the pot, which mucked everything up, and I was refluxing. Keeping it cooler worked
much better. This time, I had sieves in the condenser, heated to dissolve the glycine, then bubbled HCl through under nitrogen and let it stand
overnight. I came in to beautiful needles of the ester.
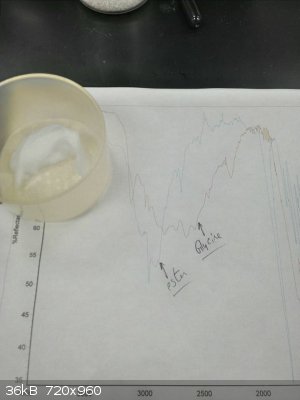
|
|
|
Dr.Bob
International Hazard
Posts: 2753
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: Mildly disgruntled scientist
|
|
Very nice job. There is another forum with many chemists and professors as well, chemicalforums.com, which is very good for interacting with other
graduate students and faculty, not that this site is not, but many of them can appreciate the situation you are in.
|
|
|
tshirtdr1
Harmless
Posts: 31
Registered: 12-12-2014
Member Is Offline
Mood: No Mood
|
|
Will check that out.
Thanks for the link. I will check out that forum.
|
|
|








