| Pages:
1
2
3 |
Boffis
International Hazard

Posts: 1903
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Rosco, there are a lot of points here so:
Yes I am assuming from the mechanism proposed by Matsuno et al (and displayed above) that the hydrogen peroxide first generate the benzohemiquinimine
and this then undergoes Micheals addition with the nitrous acid to generate the nitro phenol derivative. Matsuno claims that the there is a much
better yield in the presence of hydrogen peroxide but with their proposed mechanism I can't see anywhere else that the presence of peroxide would
benefit the reaction. If the peroxide generates the initial benzoquinimine then theoretically only one molar equivalence of nitrous acid is required
for the actual addition. Without hydrogen peroxide then about two equivalences of nitrous acid are required (depending on the exact mechanism and
stoichiometry of the initial oxidation.
I was using acetic acid as a solvent but in hindsight I would have been better to use methanol and add only a little acetic acid with the sodium
nitrite solution. Paracetamol is not soluble enough in cold water for it to be a practical solvent. As I stated in my aims for future experiments I
will use only a slight excess over the theoretical amount required to liberate the nitrous acid.
One alternative possibility is, as you indicate in your final comments, that the reaction is nothing to do with benzoquinone derivatives and Michaels
addition but a simple nitrosation and oxidation. My experience of nitrosation is that with phenols they are exceedingly rapid while the subsequent
oxidation to nitro compounds is slow or non-existent. Some years ago I posted some experiments on the reaction of nitrous acid with guanidine. In my
final analysis I discovered that the main identifiable products were nitrosoguanidine, nitroguanidine and a little urea. The only possible source of
the nitro compound was via nitrous acid oxidation of the nitroso compound. The excess of nitrous acid over the guanidine was considerable but side
reactions where important and complex.
Direct nitrosation as the mechanism would be interesting since this would explain why the hydrogen peroxide plays a role and its role would only come
into play once nitrosation had taken place. If this is the correct mechanism then the reaction scheme needs to be modified to the addition of a
stoichiometric amount of nitrite and acetic acid in the cold and then hydrogen peroxide under slightly warmer conditions. It would also explain why in
the absence of peroxide a large excess of nitrous acid is required and the yield so low as excess nitrous acid + phenol = a mess (from personal
experience). The idea of adding nitric acid afterwards is also an interesting idea to test. Once nitrosation has occurred the nitric acid should give
the appropriate nitro compound. The only question then is why the 3 position instead of, as I would expect, the 2 position.
The brown colour of my reaction mixture suggests the presence of much nitroso material but the bright yellow spherules in the initial ppt do suggest
that some nitro compound was formed but it is not easy to separate.
Somewhere in may mass of papers I have one on the synthesis of some heterocyclics containing the quinoxaline core via this reaction and it contains
excellant details of experimental conditions. I will try and find it. (Edit) Found it but it is ref 3 of Nicodems list above and it uses fuming nitric
acid so not relevant though a well described procedure.
As for experiments in stomachs or stomach like conditions they are hardly comparable to the conditions we are discussing; lower pH much lower
concentrations and and higher temperatures.
[Edited on 2-7-2015 by Boffis]
[Edited on 2-7-2015 by Boffis]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I added this as an edit to my earlier post before I realized you had replied here on a new page so I'll say what I was thinking again here.
The only way I can see the described reaction condition happening is to have a stirred slurry of paracetamol in an acetic acid / sodium acetate buffer
at pH 4, in an ice bath, to which is added in separate streams, equal drip rates of NaNO2 solution as reactant [A] and a calculated concentration and
equal volume of acetic acid as reactant [B] This scheme could maintain the pH 4 for the reaction as paracetamol in solution reacted with the 2
incoming reactant streams. As dissolved paracetamol is nitrated and drops out of solution, fresh paracetamol from the slurry will enter solution and
react, until the reaction is complete.
Calculating the solution strengths to have the reaction begin and end buffered at pH 4 is the math that needs to be done.
Actually I think the reaction may even run faster at pH 4.5 to 4.75
If it works the way I am thinking it does, I think I know how to do this buffer scheme. You make up your buffer solution of sodium acetate and acetic
acid for slurrying the paracetamol.
It should be a fairly strong buffer solution at maybe 90 to 95% saturation at 0C.
That will give you the model composition which will be the same ratio of sodium value and acetic acid value as will be needed to formulate the equal
volumes of NaNO2 solution [A] and its simultaneously added reactant [B] which is acetic acid equivalent to neutralize the sodium value of [A] plus or
minus the amount of acetic acid required to maintain the composition of the buffer solution throughout the reaction. When all the nitrous acid has
dissipated and all the reactions are complete the pH should be sitting at pH 4 at endpoint right where it started for the slurry before additions of
reactants began.
It is a constant reaction condition scheme, a continuous reaction scheme.
I would make the buffer solution and reactant [A] & [B] buffer precursor solutions almost as strong as possible like about 90 to 95% of saturation
at 0C for the "model" solution and its maintained constant "byproduct" composition of increasing volume, but kept at a constant near saturation
concentration by the incoming reactants [A] and [B].
The common ion effect is going to come into play limiting the concentration of an acetate buffer at 0C due to the lowered solubility of the sodium
value. And I'm not sure I am reading these ancient charts correctly.
But it looks like a usable buffer "model" pH 4 buffer composition could be 20.4% acetic acid / 8.2% sodium acetate trihydrate / 71.4% H2O
Then basically you calculate the equal reactant volumes for [A] and for [B] which add to form an ultimate reaction product matching that composition,
so that when all the reactions are completed the supernatant liquid has the same composition.
Bromocresol green indicator is probably about right, keeping toward the yellow range during the reaction.
I will complete the calculations for the reactants and post the values later.
The following is a kind of educated guess and reverse engineering attempt for the abbreviated and summary reaction description provided by the Chem.
Pharm. Bull. v37(5), 1422-1423; 1989 article attached on the preceding page and is the same article as was a screen shot of the reaction posted by
Boffis. I believe it is likely the Japanese chemists used a reaction scheme closely analogous to the following:
Okay I have some preliminary calculations done well enough to see that for 10 grams of paracetamol to be nitrated is going to have a finish reaction
volume of about 800 ml and given the likelihood of foaming this size batch should be run in a two liter beaker. It may be possible to compress this
reaction volume using stronger solutions. I tried to stay well away from the saturation point on the reacting solutions so byproduct salts would not
precipitate and make any foaming more complicated to manage.
For 10 grams of paracetamol to be nitrated
Here is the reactant specification for [A] 32 grams NaNO2 + 367.7 ml H2O = calculated total volume 382.5 ml
Here is the reactant specification for [B] 184.8 grams acetic acid + 206.5 ml H2O = calculated total volume 382.5 ml
The volumes should be equal for [A] and [B] or very close. Any possible small effect of solution compression or density differences between 20C and 0C
was not factored. If the specification calculation based on density is slightly off then it can be adjusted with H2O to make the volumes exactly
equal.
The initial slurry of paracetamol should be made up by adding just enough of the "model" pH 4 buffer solution made separately, which is a composition
of 20.4% acetic acid / 8.2% sodium acetate trihydrate / 71.4% H2O. Add just enough of this solution to make the paracetamol a thin stirrable paste or
thin slurry suspension.
At 0C with stirring begin simultaneous additions at equal rates [A] and [B] and the reaction should proceed buffered at pH 4.
Bromocresol green should work as an indicator to monitor pH, being kept towards yellow for the reaction to proceed. The end product is IIRC yellow
and the expected quench pH possibly about pH 5.5 due to sudden reduction of the active evolution of HNO2. The mix of color in the optimum range is
likely then to be a yellowish green with the quench arising in the really solid green that would come with rising pH in the likely pH 5.1 to pH 5.5
range with the peak HNO2 activity expected at about pH 4.8 to pH 5. Anyway, the Japanese chemists specified pH 4 so pH 4 is what I modeled, just in
case they are right on the bullseye. It could be interesting to try an alternate model with the buffer at pH 4.8 to see if yield goes up or down.

There should be adequate solubility for the paracetamol slurried in the 20.4% acetic acid / 8.2% sodium acetate trihydrate "model" buffer reaction
medium for it to very gradually dissolve enough to react incrementally in the contemplated reaction scheme.
How cleanly will be the separation of the end product is unknown, and is dependent upon the solubility in the spent reaction mixture. Neutralzing and
salting out may be required, and / or evaporation possibly vacuum evaporation at reduced temperature to facilitate recovery of the end product. Or an
extraction using a solvent, possibly methylene chloride or toluene may be required.
Attachment: Sodium Acetate Solubility Seidell.pdf (111kB)
This file has been downloaded 725 times
[Edited on 7/2/2015 by Rosco Bodine]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Nicodem  | | , you will still have to deaminate by nitrosating a p-hydroxyaniline which I do not think it is possible without ending with the
corresponding p-benzoquinone (I could be wrong though, but not motivated enough to check the literature). |
nicodem, your thinking is correct.P-aminophenols will rapidly oxidise to quinones in basic media.That's why the Orsyn paper converts the phenolic OH
into ether
really ? I read somewhere that if highly acidic medium was used(needed for a diazotisation anyway),then the oxidation of p-aminophenol to quinone can
be stopped.basically you must keep the NH2 protonated at all times to prevent the oxidation.
I remember,a long time ago, spending a lot of time thinking about making resorcinol(before I realised that the best thing to do would be to just buy
it,its so cheap).The best OTC method I could think of at that time was this:
1.nitrate benzoic acid
2.convert COOH to NH2(hoffman bromamide or schimdt),then to OH
3.reduce the nitro to amino,and then convert that to OH as well.
it seems COOH can be directly converted to OH.(a method developed by the Dow chemical company).I don't think the method is very amateur
friendly,although it has been done in a lab.the yield is low though(40%).
http://onlinelibrary.wiley.com/doi/10.1002/recl.19610800203/...
[Edited on 2-7-2015 by CuReUS]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The process described by the Japanese Chemical and Pharamceutical Bulletin article has continued to intrigue me and I have been trying to imagine an
easier way to go about reverse engineering the process described for conversion of acetaminophen (which is being assigned the acronym APAP by current
researchers) to 3-nitro APAP using sodium nitrite in a buffered reaction system where the desired reaction is reported to proceed in the range of pH
3 to pH 7. Some reports by other researchers have stated nitrosation activity is highest at about pH 5 for similar reaction schemes. And the maximum
buffer capacity for the acetic acid / sodium acetate conjugate system resides at pH 4.752 which is right in the neighborhood of the reported pH 4 by
the Japanese researchers, who also stated a range value from pH 3 to pH 7 as workable.
Some articles have stated belief that indeed a nitrosation occurs and the phenolic nitroso is then further converted to the nitro by atmospheric
oxidation. Ordinary sodium nitrite itself slowly oxidizes to the nitrate on long exposure to the air, but reportedly a phenolic nitroso is much more
greatly reactive with atmospheric oxygen and more rapidly oxidized to the nitro.
Hydrogen peroxide would likely accomplish the same oxidation much more rapidly in aqueous solution of a phenolic nitroso, converting it to a nitro.
This use of hydrogen peroxide should address the possibility that the Japanese researchers had in actuality synthesized the 3-nitroso rather than the
3-nitro compound, which seems possible since both would be reduced to the same diamino compound as was used for analytical confirmation of the product
which they obtained, which could therefore have been either the 3-nitroso or 3-nitro APAP.
Another thing that would seem relevant is that with regards to solvation of APAP there is worth consideration DMSO in which APAP is soluble to the
extent of 53% and DMSO is miscible with H2O. So it would seem an easy way to enhance solubility for APAP in H2O would be to add some DMSO and even a
small percentage of DMSO like 5 to 10% of a buffer should have a beneficial effect on the APAP solubility in the buffer.
To make up a buffer for the maximum buffer capacity pH is extremely simple for the pH 4.752 acetic acid / sodium acetate buffer at a 0.875 Molar
buffer strength.
What is required for making this buffer is to take what would be the needed total volume of buffer solution, 150 ml, as ordinary grocery store white
vinegar, 5% acetiic acid, and divide that volume into 2 equal volume portions, and add a Bromocresol Green pH indicator to one 75 ml portion, 95%
titrate it towards neutral using solid NaOH, 2.5 grams, and the remaining 5% with NaHCO3, ~0.3 gram, to complete neutralization (blue to Bromocresol
Green). Next mix the neutralized portion with the other 75 ml portion to provide ~150 ml of the pH 4.752 buffer. Two 75 ml portions of 5% white
vinegar would serve to make 150 ml of approximately 0.875 Molar buffer. To this buffer is added perhaps 25 ml DMSO and then 10 grams of APAP. The
reaction pH is monitored by the previously added bromocresol green pH indicator dye used to make up the buffer. The buffered solution begins as about
14% DMSO in which the APAP should be adequately soluble.
Reactant solution [A] would be 32 grams NaNO2 plus 50 ml H2O
Reactant solution [B] would be 47 ml of 31.45% HCl
Adjust to equal volumes of 75 ml using H2O the solutions [A] and [B]
With the APAP suspension in buffered DMSO stirring at 0C, begin adding dropwise the HCl reactant [B] until the indicator dye goes yellow, and then
begin adding the NaNO2 reactant [B] to bring the pH up and the indicator into the green. The reaction should proceed fine in the yellowish green
range, and the additions of [A] increased should increase the green, and additions of [A] decreased should result in a lowering trend on the pH
towards the yellow, while the rate of [B] remains constant.
When the additions are completed, then may be added 75 ml of 3% H2O2 or 8.5 ml of 27% H2O2 to oxidize any 3-nitroso APAP to the 3-nitro APAP.
These values for H2O2 anticipate that any 3-nitroso APAP will preferentially react with any H2O2 so that enough H2O2 will not be needed for oxidation
of all the nitrite values present in the reaction system. For that total kind of oxidation of all nitrite values would require an excessive quantity
of 3% H2O2 at 7 times the amount described and likewise for the 27% H2O2 would be applied a multiplier of 7.
If the "virtual lab" in my mind is on the bullseye with the theory aspects, what I have described should work. Isolation of the product would depend
on the solubility of the 3-nitro APAP which is unknown. Salting out, or evaporation and/or extraction could be required unless fortune favors with a
low solubility for the 3-nitro APAP which hopefully may simply crystallize out in the cold.
[Edited on 7/4/2015 by Rosco Bodine]
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
That's an interesting route. I never considered that, so I can't really comment.
I used to be interested in resorcinol many years ago. I forget exactly why, but nevermind that. The best route I could come up with while be
relatively possible for an amateur was not a clean one.
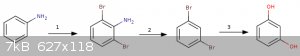
Starts with aniline then
1) EAS with Br2. No clue how well the selectivity for the di-ortho (relative the amine) would be. Pretty sure aniline is so activated here
that this would go without catalysis. If Para was preferred then huge blunder.
Edit-
Come to think of it this would likely fail to give any selectivity for the di-product especially not the right regioisomers unless the para position
was blocked. Woops.
2) Then the classy sandmeyer elimination type reaction which has been discussed here several times in several ways. Again, messy, but doable.
3) Dihydroxylation reaction not sure how clean that'd go either. I know some users have reported hardship with similar reactions.
[Edited on 5-7-2015 by smaerd]
Maybe there's a selective way to nitrate an acetanilide amide for the meta regioisomer. Or go for dinitrobenzene...
The real difficulty is just meta positioning.
edit again - just checked the wiki. It says melts of sulphonic acids are typically used. That's definitely approachable.
[Edited on 5-7-2015 by smaerd]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Aniline actually nitrates primarily to the 3<sup>rd</sup> position due to the -NH<sub>3</sub><sup>+</sup>, the
corresponding acidic cation of aniline in the typical conc. sulfuric acid solvent, being deactivating, although there will be some contamination with
the ortho and para isomers. Alternatively, nitrating benzamide yields a pure 3-nitrobenzamide, which upon a Hofmann rearrangement and reduction of
the nitro group, yields m-phenylenediamine which can be diazotised to yield resorcinol. This isn't a cost efficient route, but I am considering using
it to synthesize 3-aminophenol, I'm trying to decide if it is simpler to just use the ammonolysis of resorcinol though.
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
@Gdflp - I didn't know that. I always assumed it would nitrate ortho/para but now that you explained it that makes sense. Hmmm..
Here's a scheme that should actually be valid. Only missing one reference.
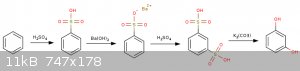
1. Start with benzene and sulfonate as usual with H2SO4.
2. Create the Barium salt with BaOH or similar.
3. Reflux with Conc Sulfuric acid at 209*C ~4Hr's to create the 1-3 disulphonate.
Benzenedisulfonic Acid from Benzenemonosulfonic. C. E. Senseman. Ind. Eng. Chem., 1921, 13 (12), pp 1124–1126. DOI: 10.1021/ie50144a012 (http://pubs.acs.org/doi/abs/10.1021/ie50144a012?journalCode=...)
4. Melt in potassium carbonate (missing ref?) Meyer, J (1897). Ber 30: 2569
Probably not cheap but it seems like there would be appreciable control in the intermediates synthesis (no regioisomer hell). Pretty simple chemistry.
Not sure what temp the melt is performed at though.
[Edited on 5-7-2015 by smaerd]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
There is a procedure for the alkali fusion of the 1,3-disulphonate on p.144 of Fundamental Processes of Dye
Chemistry in the forum library, I believe that Magpie did some experimentation with the preparation of resorcinol this way a while back.
That's a very interesting reference for the disulfonation, I was always under the impression that the disulfonation of benzene required at least 20%
oleum, such as the procedure preceding the alkali fusion in Fundamental Processes of Dye Chemistry.
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Come to think of it, I think I read Magpie's synthesis a year or so ago! Or maybe it was a napthalene derivative(http://www.sciencemadness.org/talk/viewthread.php?tid=11663#...)? Either way, same type of chemistry.
Supposedly, the publication: Polak. Rec. Trav. Chim. , 29, 1910,
States that the barium salt in 98% acid (sulfuric) is sufficient. Then again, maybe in 1910 industrial acid had high concentrations of oleum. The
reason I believe it wouldn't is that they state 98%. Could be a load of shit though.
[Edited on 5-7-2015 by smaerd]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2894
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
From aniline nitration with conc HNO3 there is 50/50 mix of p-nitroaniline (+traces ortho) and m-nitroaniline.
I would start from benzene (going a bit in the same direction as CuReUS)
1°)dinitration to get 1.3-dinitrobenzene.
2°)1,3-DNB is partially reduced with NH4SH or Na2S/H2S (beware on stoechiometry to avoid reduction of the secon nitro group) to get 3-nitroaniline
3°) 3-nitroanilinine is diazotized with H2SO4/NaNO2 and reacted with water/ethanol upon boiling to give 3-nitrophenol
4°) 3-nitrophenol is reduced by NH4SH or Na2S/H2S or Fe/HCl or SnCl2/HCl to get 3-aminophenol
5°) 3-aminophenol is diazotized with H2SO4/NaNO2 and reacted with water/ethanol upon boiling to give resorcinol.
Some precautions must be taken to avoid copulation of the 3-amino-phenol with the diazocompound (the aminophenol must fall into the nitrosating bath
(in exces thus) sothat at any moment the aminophenol reacts immediately with HNO2 and not with the diazo-compound.
The total reduction of the DNB into 1,3-diaminobenzene is possible and bis-diazotation also following certain rules to avoid copulation of the
diazocompound with the diamine to form Bismark Brown.
The process of smaerd with benzodisulfonic acid and base fusion (usually NaOH) is a good one.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2894
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
@Rosco and Boffis,
The nitrosation of paracetamol (para-acetaminophenol) is a stange story!
In books I see that:
-sometimes nitrous acid provoques the deacetylation and diazotation of the amino group
-sometimes there is reference of acetanilide being nitrosated as N-nitroso-acetanilide (isolable) wich upon basification (KOH) turns into K acetate
and K benzodiazotate.
-phenols tends to nitrosate in para to the hydroxy group, but if not accessible then it goes to ortho position to the hydroxy group...why then the N=O
in position 3 instead of 2...does it come from a transcient N-Nitroso-p-acetamino-phenol and vicinal deplacement at the closest position?
-nitrosoaromatic may oxydise with HNO2 into nitroaromatics
About bromocresol green...is it stable towards nitrous acid?
The colour shift is interesting but if you form a yellow compound in the blue shift region the total solution will look green giving misleading info
about pH.
If you form a brown compound the colour will also be hard to figure...
Last but not least...paracetamol will also make a buffer arround pH 9.3 +/-1 and the resulting 3-nitroparacetamol will add complexity with a third
buffer arround pH 8.6 +/-1...
So wich one will win? Acetic acid/acetate, p-acetaminophenol/p-acetaminophenate or 3-nitro-p-acetaminophenol/3-nitro-p-acetaminophenate?
I think you are only at the start of a very hard work 
This may help as it is related (see S11 to S20)
Attachment: Functionalized alkoxy arene diazonium salts from Paracetamol.pdf (5.3MB)
This file has been downloaded 829 times
[Edited on 5-7-2015 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Boffis
International Hazard
Posts: 1903
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Rosco. I’ve re-read your post above and thought about this for some time. I was not really happy with my own initial attempt, however, a few points
are worth making before I discuss your proposals. In the original Japanese paper they stated a 3 to 5-fold excess of nitrous acid between pH 3 and 7.
I made no attempt to buffer the reaction mixture, the large amount of acetic acid was merely to act as a solvent for the paracetamol but the pH would
still have been >3 and so within the lower limit, admittedly only just. I agree that a buffer to give a pH closer to the mid-range would have been
better but this was intended to be the first of a series of experiments. Unfortunately I have been sent overseas again and will not be home for
another 6 weeks.
Thinking about things again the large excess on nitrous acid make no sense. My experience of phenolic nitrosation reactions is that they are very
rapid and the subsequent oxidation is slow. So with the addition of a more potent oxidizing agent the addition of more than one equivalence of sodium
nitrite is un-necessary. In fact my experience is that excess nitrous acid almost always results in the generation of much tarry material in any
reaction and my experience, as posted above, with this reaction seems to bear that out.
Looking at your proposals I could carry out these reaction as prescribed but I have a few issues:
1) Even without the addition of hydrogen peroxide your sodium nitrite addition seems vastly excessive. For 10g of paracetamol a threefold excess would
be only 13.7g and a fivefold excess 22.8g or were you working on the 7-fold equivalence (which I don’t understand) you mentioned about 3 post back?
In the original paper they state that the yield of 22% was obtained with the excess of nitrous acid and with hydrogen peroxide the yield was 94% but
now they don’t state that they used an excess of nitrous acid at all and I believe they didn’t and were being deliberately obscure. The authors
merely state that the nitration is “greatly facilitated by treatment with equimolecular amounts of hydrogen peroxide…”.
2) Given that any concentration of acetic acid will give you pH of 3-7 I think we are going overboard on pH buffering. I think that to use a
suspension of finely ground paracetamol as you suggest or with a small volume of co-solvent such as methanol or DMSO it will be sufficient to add the
sodium nitrite and acetic acid with a slight excess of the latter. A small quantity of acetic acid may be added to the paracetamol solution/suspension
just to ensure a starting pH of <7. I am always wary of swamping the reaction with dissolved salts unnecessarily.
My plan is to start out with a 1:1 ration of paracetamol and nitrous acid at pH >4 and see if I can isolate the 3-nitroso compound and then see if
I can oxidise this with hydrogen peroxide to the nitro compound. I will then repeat the experiment changing the various parameters until I get
products that appear reasonable. As I stated in my initial experimental details the initial precipitate contained both a yellow and a brown product
while the subsequent ether extraction was crystalline and orange with a little tarry material so it looks like I got a mixture of products but the
nitro compound is most likely the yellow one as the nitroso compounds are usually brown.
By the way I found a paper that describes the assay of paracetamol in blood serum using the nitration with nitrate and they claim that the product is
the 2-nitro compound but they carry out the nitration in trichloroacetic acid-hydrochloric acid so probably lower pH but the important thing is that
the reaction appears almost instantaneous.
The next question is how do we know what we have got? My plan now is to try the reaction above with only one equivalence of nitrous acid and then try
reducing the nitroso compound to and amine. Then without de-acetylation attempt to diazotize the product with one equivalence of nitrous acid to see
if I can produce 1-acetyl-5-hydroxybenzotriazole directly. I have a paper (somewhere) that gives the properties of the 5-hydroxybenzotriazole.
I also intend to try the di-acetylation with acetic anhydride and then nitrate to provide material for comparison.
I also intend to test the various products with either 4-amino-dimethylaniline or sodium sulphanilate. Many aromatic nitroso compounds condense with
these amines to give azo dyes while nitro compounds do not normally react. Possible removal of excess nitrous acid may be necessary first with urea or
similar. I am going to investigate the product from the initial experiment with chromatography (TLC on silica 60 plates any ideas about solvents?) to
see how many compounds are present. Another test might be ferric chloride which gives a deep green colour with most 2-nitrosophenols so this test
would be a negative test in that the 3-nitroso compound should not.
@smaerd, I don't think that brominating aniline would work like that. I have a reference somewhere to bromination aniline with N-bromosuccamide to
avoid oxidation and side reaction IIRC.
@Philou, Nitrosation of paracetamol strange! that's an understatement. Part of the problem though is some of the papers had only a passing interest in
the outcome so didn't persue the product while others are just vague.
Yes I think the colour of the reaction mixture will mask the colour of bromocresol green if used in the reaction mixture (and survives) I would use
the dye to prepare paper strips. Actually all of the reactive (ie phenolic) o and p sites in bromocresol green have already been blocked so the
compound may survive under the mildly acid conditions, it depends on its ability to resist oxidation.
And, as for all those buffers; my brain hurts . I think I stick with my system
given above because the addition of sodium nitrite will have its own buffering effect too. . I think I stick with my system
given above because the addition of sodium nitrite will have its own buffering effect too.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by Boffis | | @Rosco. I’ve re-read your post above and thought about this for some time. I was not really happy with my own initial attempt, however, a few points
are worth making before I discuss your proposals. In the original Japanese paper they stated a 3 to 5-fold excess of nitrous acid between pH 3 and 7.
|
No, what they explicitly stated was use of 2 moles NaNO2 for the theoretical reaction plus a 5 moles NaNO2 in excess of theory gave an 81% for the
reaction at pH 4.
| Quote: |
verbatim from the article:
Thus, p-acetamidophenol [1] was treated at OC with a five-molar excess of sodium nitrite in aqueous acetic acid at pH 4 or in a phosphate-buffer
solution at pH 7 to give the 3-nitrated phenol [3] (m p139C) in 81% yield. |
The use of hydrogen peroxide suggests that the nitroso rather than the nitro is what has been obtained in 81% yield.
A similar thing can be done with nitrosoresorcinol which can be oxidized to the nitrate by hydrogen peroxide, or by HNO3.
| Quote: |
I made no attempt to buffer the reaction mixture, the large amount of acetic acid was merely to act as a solvent for the paracetamol but the pH would
still have been >3 and so within the lower limit, admittedly only just. I agree that a buffer to give a pH closer to the mid-range would have been
better but this was intended to be the first of a series of experiments. Unfortunately I have been sent overseas again and will not be home for
another 6 weeks.
Thinking about things again the large excess on nitrous acid make no sense. My experience of phenolic nitrosation reactions is that they are very
rapid and the subsequent oxidation is slow. So with the addition of a more potent oxidizing agent the addition of more than one equivalence of sodium
nitrite is un-necessary. In fact my experience is that excess nitrous acid almost always results in the generation of much tarry material in any
reaction and my experience, as posted above, with this reaction seems to bear that out. |
What are the reaction conditions are not stated for concentration or for the time required. So the article is cryptic about important details.
| Quote: |
Looking at your proposals I could carry out these reaction as prescribed but I have a few issues:
1) Even without the addition of hydrogen peroxide your sodium nitrite addition seems vastly excessive. For 10g of paracetamol a threefold excess would
be only 13.7g and a fivefold excess 22.8g or were you working on the 7-fold equivalence (which I don’t understand) you mentioned about 3 post back?
|
Yes 1 mole NaNO2 for oxidizing the APAP to the imine plus 1 mole NaNO2 for nitrosation of the imine plus a 5 molar excess which is what the article
stated was used. 1 + 1 + 5 = 7
| Quote: |
In the original paper they state that the yield of 22% was obtained with the excess of nitrous acid and with hydrogen peroxide the yield was 94% but
now they don’t state that they used an excess of nitrous acid at all and I believe they didn’t and were being deliberately obscure. The authors
merely state that the nitration is “greatly facilitated by treatment with equimolecular amounts of hydrogen peroxide…”. |
We can agree the authors are being obscure or certainly there is much lost in translation. Also the article states that more information related to
the synthesis was provided at the annual meeting of the Pharmaceutical Society of Japan, but I have not been able to find the Abstracts for that
annual meeting, which are likely not translated anyway.
That relates to the imine isolated separately not necessarily to the reaction shown where the imine is further reacted in situ.
It seems like it should apply equally well but not necessarily if the excess of HNO2 was being used as an oxidizer instead of using H2O2 in tandem and
subsequent to the HNO2, which could speed up the oxidation and reduce the HNO2 requirement. I believe a 3-nitroso APAP is actually what is gotten as
a second intermediate but is not identified by the reactions given by Matsuno et al.
| Quote: |
2) Given that any concentration of acetic acid will give you pH of 3-7 I think we are going overboard on pH buffering. |
Controlling the pH may govern the 3 position being preferred by substitution. It may also govern the oxidation activity for the HNO2.
| Quote: |
I think that to use a suspension of finely ground paracetamol as you suggest or with a small volume of co-solvent such as methanol or DMSO it will be
sufficient to add the sodium nitrite and acetic acid with a slight excess of the latter. A small quantity of acetic acid may be added to the
paracetamol solution/suspension just to ensure a starting pH of <7. I am always wary of swamping the reaction with dissolved salts unnecessarily.
My plan is to start out with a 1:1 ration of paracetamol and nitrous acid at pH >4 and see if I can isolate the 3-nitroso compound and then see if
I can oxidise this with hydrogen peroxide to the nitro compound. I will then repeat the experiment changing the various parameters until I get
products that appear reasonable. As I stated in my initial experimental details the initial precipitate contained both a yellow and a brown product
while the subsequent ether extraction was crystalline and orange with a little tarry material so it looks like I got a mixture of products but the
nitro compound is most likely the yellow one as the nitroso compounds are usually brown.
By the way I found a paper that describes the assay of paracetamol in blood serum using the nitration with nitrate and they claim that the product is
the 2-nitro compound but they carry out the nitration in trichloroacetic acid-hydrochloric acid so probably lower pH but the important thing is that
the reaction appears almost instantaneous.
The next question is how do we know what we have got?
|
See page 10 of the pdf attached, page 442 of the journal article. The melting point of 218C is different from the 139C reported by Matsuno et al so
this is more evidence they have the 3-nitroso rather than the 3-nitro compound.
If the F. 218 identified by Reverdin is fahrenheit, there is still a discrepancy because that is 103 C which is not the 139 C given by Matsuno. There
is a problem there with the mp either way.
You and I have found errors before in the literature, haven't we? Keeping all
those phd's honest is becoming tedious work. Maybe they suffer from confirmation bias writ large.
| Quote: |
My plan now is to try the reaction above with only one equivalence of nitrous acid and then try reducing the nitroso compound to and amine. Then
without de-acetylation attempt to diazotize the product with one equivalence of nitrous acid to see if I can produce 1-acetyl-5-hydroxybenzotriazole
directly. I have a paper (somewhere) that gives the properties of the 5-hydroxybenzotriazole.
I also intend to try the di-acetylation with acetic anhydride and then nitrate to provide material for comparison. |
Getting down to brass tacks, a la Mendola and Reverdin
| Quote: |
I also intend to test the various products with either 4-amino-dimethylaniline or sodium sulphanilate. Many aromatic nitroso compounds condense with
these amines to give azo dyes while nitro compounds do not normally react. Possible removal of excess nitrous acid may be necessary first with urea or
similar. I am going to investigate the product from the initial experiment with chromatography (TLC on silica 60 plates any ideas about solvents?) to
see how many compounds are present. Another test might be ferric chloride which gives a deep green colour with most 2-nitrosophenols so this test
would be a negative test in that the 3-nitroso compound should not.
Yes I think the colour of the reaction mixture will mask the colour of bromocresol green if used in the reaction mixture (and survives) I would use
the dye to prepare paper strips. Actually all of the reactive (ie phenolic) o and p sites in bromocresol green have already been blocked so the
compound may survive under the mildly acid conditions, it depends on its ability to resist oxidation.
And, as for all those buffers; my brain hurts. I think I stick with my system
given above because the addition of sodium nitrite will have its own buffering effect too.
|
Hey the buffers are easy at the pKa they all work the same
And a java applet can do the rest.
http://clymer.altervista.org/buffers/acetic.html
Here's another article attached that shows H2O2 used to oxidize dinitrosoresorcinol probaly the 2,4-dinitroso to dinitroresorcinol See page 8 of the
pdf
There is a patent for this too I think. The reference for the H2O2 use is not stated or cited, so I'll have to look for it.
You don't want to use methyl alcohol for a cosolvent because of easy formation of methyl nitrite which is like ether or butane in volatility and will
escape with loss of nitrogen from the reaction system. If methanol was used as a cosolvent by Matsuno that could easily explain the need for a 5
molar excess because most of the needed nitrogen in the reaction mixture just like Elvis had left the building.
Solubility for APAP at OC is .72 grams in 100 ml H2O so really a cosolvent is probably not needed, and even if DMSO was added it wouldn't take very
much maybe 2% DMSO to triple the water solubility and that solubility could be the reaction rate governor. However there is a possibility that
changing the solvent system could also change the product. For example when resorcinol is treated with nitrite in aqueous medium a nitroso derivative
results. But when treated in solution in ether a diazo derivative results. A similar difference could occur by changing the solvent system with
APAP.
Matsuno cited these articles (reference 7) with regards to the imine being isolated and reacted separately
http://pubs.acs.org/doi/abs/10.1021/jm00350a001
http://www.sciencedirect.com/science/article/pii/S0040403900...
Here is another article that could be interesting
http://pubs.acs.org/doi/abs/10.1021/ja01659a030
Attachment: Reverdin and Dresel, Archives des sciences physiques et naturelles, 1904, 18, 442.pdf (392kB)
This file has been downloaded 688 times
Attachment: AD0399653.pdf (1.8MB)
This file has been downloaded 741 times
WRT the bromocresol green if it does survive the reaction system then yes I knew the green range would likely be obscured, but thought the yellow
would probably work as the marker. What will be the color development for the reaction mixture is an unknown that could defy any use of any indicator
dye and it could require monitoring with a meter. Also possible is that any buffer system would be overloaded but that would tend to not make sense
for reason that use of a buffered reaction system was specified by Matsuno. To specify a buffered system if a buffer wasn't even used would be
misleading more than simply being deliberately obscure and would be unethical and dishonest.
As a note on the subject of pH indicator dyes, I ran across this interesting bit about lacmoid or lackmoid (resorcinol blue) pH indicator

Attachment: lackmoid pH indicator Bulletin_of_Pharmacy Vol2 1888 pg286.pdf (278kB)
This file has been downloaded 648 times
[Edited on 7/6/2015 by Rosco Bodine]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by PHILOU Zrealone |
The total reduction of the DNB into 1,3-diaminobenzene is possible and bis-diazotation also following certain rules to avoid copulation of the
diazocompound with the diamine to form Bismark Brown. |
I later realised that it would be better to do a double diazotisation on 1,3-diaminobenzene instead of doing it one by one because diazo reactions are
very messy.Mix the diamino with NaNO2+HCl in a sep funnel and drip it into a beaker of boiling water with a few drops of acid.
can you tell more about the rules on bismark brown .
| Quote: | | The process of smaerd with benzodisulfonic acid and base fusion (usually NaOH) is a good one. |
No,it isn't.It looks very easy, but its not.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
@ Boffis and Rosco:
The paper regarding nitrite mediated nitration of acetaminophen to the supposed 3 nitro 4 acetaminophenol is very sparse with detailed information
indeed. Interesting idea regarding nitroso-nitro oxidation instead of the proposed mechanism by the authors.
Here is my experience with the reaction: I've attempted this reaction only once and on a very small scale of 1 gram. Basically, I simply suspended 1
gram of finely powdered purified acetaminophen in 15 ml 10% AA, and added dilute NaOH solution untill the pH was 4.5-5.0. While sitrring in an ice
bath, 1 gram of nitrite solution was added at once. The nitrtion proceeded very slowly, going from colourless to a bright orange colour over the
course of a few hours. I did't attent the whole reaction, but a sort of induction point seemed to be present, after which the reaction proceeded
faster and with some foaming. End product was voluminous bright orange powder. The colour very much resembles DNAc. Attempted deacetylation suing 50%
SA resulted in brown crud.
[Edited on 9-7-2015 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Instead of deacetylation it would have been interesting to add some HNO3 and see if the bright orange powder might then change color or form.
|
|
|
Boffis
International Hazard
Posts: 1903
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@nitro-genes. Thank you for your contribution, it is actually rather interesting as you used a lower concentration of acetic acid that I did and
therefore a higher pH and it sounds like you got a cleaner product but again yours sounds like a nitroso compound too. Nitroso compounds tend to be
orange-brown were as the equivalent nitro compounds tend to be yellow or occasionally red (interesting o-nitroaniline, a closely related compound to
our 2-nitro-4-hydroxy-acetanilide, is red).
A feature of nitroso compounds is there tendency to turn into brown crud in strongly acid conditions (in my experience which includes a lot of such
material!) so again it sounds to me like we are getting largerly nitroso compounds.
When I did this I made a homogeneous solution but this took a lot more acetic acid but the reaction was immediate and appeared rapid and effervescence
only occurred after the addition of one molar equivalence of nitrous acid. The initial precipitate was an obvious mixture of a bright yellow
crystalline "ponpoms" and an amorphous looking dark plain-chocolate brown material. I only got an orange crystalline substance when I extracted the
reaction mixture with ether.
As I say when I get home again I'll investigate the products further and also repeat the experiments under less acid condition and see if I can
isolate the intermediates. I agree entirely that the original paper lacks details and little compelling evidence that a nitro compound was produced.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
@nitro-genes and @Boffis
There are some very interesting properties for paracetamol as a reagent having easy reactivity which allows for its subsequent reactions along
different paths leading to other interesting compounds. Several reaction paths are possible which lead to good yields or even complete conversions of
the paracetamol to another compound.
One of the things I learned is that paracetamol is itself a reducing agent and will reduce ferric iron to ferrous iron while the paracetamol is itself
converted to a quinone, and I think how far the oxidation proceeds is governed by temperature and probably by pH. The oxidation product may even
complex with the reduced ion of the metal. I think possible products of further oxidation may be N-acetyl-P-benzoquinone imine or p-aminophenol or
hydroquinone or benzoquinone, and that paths for derivatives of each of those can be optimized so that one desired product predominates.
There have been discussions before about the interesting reactivity of paracetamol.
http://www.sciencemadness.org/talk/viewthread.php?tid=9424 chloranil
http://www.sciencemadness.org/talk/viewthread.php?tid=8250#p... benzoquinone
http://translate.google.nl/translate?js=y&prev=_t&hl...
Another intriguing idea is that a sequence of reactions could occur where the paracetamol could be partially oxidized and converted to a 3-nitroso
under mild conditions using a nitrite, possibly a ferric nitrite? maybe ferric chloride plus sodium nitrite, subsequently there is used nitric acid to
oxidize the 3-nitroso to a 3-nitro. If deacetylation subsequently occurred possibly via HCl or H2SO4, the result would be 3-nitro-p-aminophenol,
which could be both diazotized and nitrated with raising the H2SO4 to about 25-30% in the reaction mixture, with another dosing of NaNO2 to provide
the conditions described by Benedikt and Hubl, possibly producing an unknown? isomer of DDNP as the result, which could be a 4-diazo-3,6-dinitrophenol
or a 4-diazo-3,5-dinitrophenol.
If such a reaction scheme is possible through a stepwise sequence of manipulations of mild reaction conditions, it would be pretty unique for
production of an energetic material of that potential energy under such mild reaction conditions. And it might be possible as a "one pot" synthesis.
Another interesting possibility is that diazotization in the presence of perchlorate ion can in some cases lead to a precipitation of an unstable
transitional intermediate low solubility diazonium perchlorate salt, which may itself have interesting energetic properties.
[Edited on 7/9/2015 by Rosco Bodine]
|
|
|
clearly_not_atara
International Hazard
Posts: 2836
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
NAPQI is sort of interesting, but it's worth noting that its hydrolysis products, benzoquinone and acetamide, are both pretty useful in and of
themselves. I doubt you'll find any literature trying to recover acetamide from NAPQI hydrolysis, but at the same time I'm sure it's not that hard.
But more interesting is that N-nitroso-N-arylacetamides are converted to phenol methyl ethers by reaction with MeOH / H+:
http://pubs.acs.org/doi/abs/10.1021/ja01148a013
While NaNO2 will oxidize paracetamol, O-alkylparacetamol will not be so susceptible to oxidation, and the expected product of methanolysis on
N-nitroso-O-methylparacetamol is p-dimethoxybenzene. Formylation of O-alkylparacetamol may be possible and gives a more interesting methanolysis
product...
[Edited on 10-7-2015 by clearly_not_atara]
[Edited on 10-7-2015 by clearly_not_atara]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Evidence accumulates that the alleged 3-nitro-acetaminophen of Mansunto et al is NOT the 3-nitro but is almost certainly the 3-nitroso-acetaminophen
which would have reduced to the same diamino compound which was the basis for identification of the likely precursor. Here is an abstract from the
time when different identifications of bona fide 3-nitro-acetaminophen were made by different synthetic methods and the mp identified at 218C

Attachment: Spectrophotometric determination of acetaminophen and salicylamide through nitrosation and subsequent chelation.pdf (167kB)
This file has been downloaded 1013 times
See also the attached article published 10 years before the Mansunto et all article which describes a nitrosation of acetaminophen under alkaline
conditions as producing a C-nitroso derivative at the 2 position with concurrent dehydration of the phenol to the quinone.
From the references which have been reviewed thus far, it appears to me that the nitrosation or "nitritation" of acetaminophen may proceed different
ways, depending upon the pH and probably the temperature also, so that the result may be a conversion of the hydroxyl to the oxime, or an alternate
outcome may be a C-nitroso at position 2,(possibly an oxime interactive with the phenol hydroxyl), or a third outcome may be a C-nitroso at position
3,(possibly an oxime interactive with the N-acetyl or with the phenol hydroxyl), and I think a fourth possible outcome may involve a reaction with the
4 position N-acetyl group,(possibly an oxime interactive with the phenol hydroxyl). And while those would seem to be the main 4 possible reactions,
there doesn't seem to be any clear mapping of the reaction paths and outcomes ever charted as a kind of summary in any publication describing the
synthetic conditions and the various yields and byproducts. In their totality it appears these various reactions are simply not yet charted, thus we
are in "uncharted territory" .....like going where none have gone before Who
would have guessed
Anyway, sufficient to say that Mansunto et al, have just been sent back to school. And if the structural identifications have been based upon NMR
"library" data then obviously the NMR library data is WRONG also.
Nicodem I think recognized and alluded to this possibility when making note earlier that reduction of a nitroso or a nitro could lead to the same
amino as the result, so it is not conclusive to identify a nitro as positive identification of what was the actual precursor for the amino. It is a
coin toss 50/50 chance it was one or the other, and in this case Mansunto et al, simply guessed WRONG on the coin toss and did not follow up to
positively confirm which precursor for the amine had actually been reduced. Evidently it was indeed the 3-nitroso and not the 3-nitro as they
presumed.
A classic example would be the case of aminoguandine, where either nitroguanidine or nitrosoguanidine are both reduced by hydrazine to the same
aminoguanidine. The nitroso is an actual identified intermediate in the case where the nitro is being reduced to the amino compound. A similar
scenario is occurring here with the Mansunto et al article. They presumed the precursor for the amine would have been the nitro being reduced to the
amine, but it was actually the nitroso which they reduced to the amine.
This is a discovery I had actually anticipated when I posted my thoughts earlier
| Quote: |
What is going to be the potential "real bite" here is if the Japanese have misidentified a nitroso for a nitro.
3-nitroso instead of 3-nitro
would not make me a happy camper
If indeed it does turn out to be a 3-nitroso then it may possibly oxidize to the nitro by warming with dilute HNO3 |
It's deja vu all over again I was on the bullseye.
Can't fool old Rosco Trying to pass off a nitroso as a nitro ....shame ....shame
Mansunto et al ........You are so busted!!!
You have the right to remain silent, anything you say can and will be used against you in a court of peer review If you cannot afford a chemist .....one will be appointed for you
https://www.youtube.com/watch?v=ja0_m-4NAec
<iframe sandbox width="640" height="480" src="https://www.youtube.com/embed/ja0_m-4NAec?rel=0" frameborder="0" allowfullscreen></iframe>
[Edited on 7/11/2015 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Victory lap ^^^^^^^^^^
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
There was an interesting post in this forum 5 years ago which should receive follow up in connection with this discussion, even though it identifies
probable 2-nitroso or 2-nitro acetaminophen.
http://www.sciencemadness.org/talk/viewthread.php?tid=5617&a...
| Quote: | | Sodium Nitrite and HCl are added to an Aq. Solution of acetaminophen, to give (N-acetyl)-4-hydroxy-3-nitroaniline |
There is a link by the poster to this page which is a subscriber only restricted access
http://www.chemedx.org/jce-product/chemistry-comes-alive
Does anyone here have a subscription and login who can help with providing that content?
Another relevant thread is here
http://www.sciencemadness.org/talk/viewthread.php?tid=27849&...
[Edited on 7/14/2015 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Here is another paper related to the topic of nitration of acetaminophen but this paper reports a nitration using nitrite which provides the
2-nitro-4-acetylaminophen synonymous with 3-nitro-4-hydroxyacetanilide, reported mp 180C with two references
This is likely the result of a more acidic reaction system than the buffered slightly acidic reaction condition which was used by Manusnto et al to
produce the alleged 3-nitro acetaminophen believed to more probably be the 3-nitrosoacetaminophen reported by Mansunto as mp 139, compared to
authentic 3-nitroacetaminophen mp 218C reported by Reverdin.
Attachment: molecules-07-00734 3-Nitro-4-Hydroxy-Acetanilide.pdf (43kB)
This file has been downloaded 802 times
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
@Boffis It has been pretty well established that the compound reported by the Japanese as the 3 nitro was actually the 3 nitroso acetaminophen. So
the use of H2O2 would likely be applied subsequently to the nitroso product as an increase over the speed of an air oxidation of the nitroso to the
nitro. Some of the nitroso derivatives will oxidize to the nitro on long standing exposure to air or being aerated in solution. I am thinking the
more active oxygen of H2O2 would speed that process. Nitric acid would probably work as well and maybe better to convert the nitroso to the nitro.
There is precedent for conversion of such nitroso groups to nitro groups by use of nitric acid. It may require some heating also.
[Edited on 2/27/2017 by Rosco Bodine]
|
|
|
Boffis
International Hazard
Posts: 1903
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Rosco, I am going to have another go at this reaction but my paln is to suspend the the paracetamol, sodium nitrite and sodium acetate with and
without H2O2 in a mixture of methanol and water and then slowly add slightly more acetic acid than required to liberate the nitrous acid. This should
keep the pH fairly high, close to 7 to start with. My theory is that if the reaction is too slow at 7pH then the free acids will cause the pH to fall
until reaction occurs. I am working on the quantities at present.
|
|
|
| Pages:
1
2
3 |








