| Pages:
1
2
3
4
5
6
..
26 |
Eclectic
National Hazard

Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Thanks, my bad. :-)
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Increasing yield..
It would be good if it were possible to extract the benzaldehyde out as it forms, it would certainly increase the overall conversion.
But, if it is possible to get a near total conversion using simple means, then it won't be necessary to extract out the benzaldehyde. Perhaps
stirring will result in a much higher conversion.
I will find out soon enough, this evening I distilled out the cinnamaldehyde from the rest of the cinnamon oil. Tomorrow, I will add the other 128g to
the 300 ml (I haven't weighed it yet) I've just distilled.
What do you folks think will happen if the dilution is decreased?
I'm considering reacting the roughly 430g I have left, in one reaction in a 3 L RBF. If I fill the 3 L RBF with 2.5 L of liquid, then that is a
dilution of approx. 40%.
I can also let it react for a few hours longer, I don't yet have any reason to think that would do any harm.
Any ideas?
Here are a couple photos I took of the cinnamon distillate earlier this evening:
http://www.sciencemadness.org/scipics/cycloswirlies.jpg
http://www.sciencemadness.org/scipics/cycloswirlies2.jpg
[Edited on 7-2-2005 by CycloKnight]
|
|
|
Eclectic
National Hazard
Posts: 899
Registered: 14-11-2004
Member Is Offline
Mood: Obsessive
|
|
Use a surfactant and stir. More surface area between cinnamon oil and water should speed up conversion. Use hot water for reflux if you don't
want to try the steam azeotrope distillation idea, as that will at least allow the acealdehyde to escape.
If you use a distillation column and distill off the benzaldehyde as if forms, you won't need to do an extraction or distillation later. You
might need some way to return the upper water layer to the boiler automatically though. I don't have the composition of the steam/benzaldehyde
at hand, and it could be a lot of water for a small amount of product.
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Benzaldehyde
If all goes well, I should have the last batch of cinnamaldehyde (to benza.)started this evening.
I'm leaning towards carrying it out using the higher concentration of sodium carbonate that I mentioned before. That would really speed things
up. Could you imagine processing 3 litres of cinnamon oil? That would require a 7 hour reflux for 30 litres! With good mixing remember..
With these new concentrations, it would only require around half of that, not too bad.
I'm very much looking forward to see what kind of yields can be expected from now on using this more concentrated variation.
Things would be more scientific if I were to do exactly what I did before except this time apply the improvements I mentioned. I intend to apply those
improvements AND using the higher concentration (less water).
If things turn out bad, it could be difficult to narrow down the cause.
But, I'm in a rather optimistic mood - go for gold shall I?
If anyone can see any reason why I could end up ruining all of my remaining cinnamaldehyde based on the changes I've proposed, please speak now,
or forever hold your peace...
I'm going to use:
1) approx. 430g cinnamaldehyde
2) 125g NaCO3
3) >2L of boiled water
If I use 2 L of water, then I've more than doubled the concentration from before.
Shall I start with say, a 10 hour stirred reflux? Or perhaps I should increase it to 14?
Magnetic stirring will be as rapid as I can get it, as long as the stir bar remains stable.
I dare say, that it may be possible to increase the conversion rate, by adding a solvent to help dissolve the cinnamaldehyde (or the original oil).
Such as 10% ethanol?
I won't do that this time though, too many unknown variables.
Maybe next time, depending on the yield I can manage this time.
Now on to the toluene oxidation.
Just as a reminder to those just joining this page, MAA = manganese-ammonium alum, and MAS = manganese-ammonium sulphate. MAA is the dark red
oxidizer.
Yellow MAS is converted to dark red MAA in the acid cell when current is applied.
The toluene oxidation cell experiment is still ongoing, and will be for a while. However, I've been observing some rather bizaare things going
on. I don't yet have any pictures of this. I am for the first time, now starting to find oxidizer masses growing on the cathode, this happened
when the cathode was submerged in MAS silt. I have also observed little masses of oxidizer growing on the sides of the beaker...
Also, when the "anodemites" are allowed to form when STIRRING has been applied, the unoxidized silt gets "cemented" into the
precipitating oxidizer as well, to form a wierd, solid conglomerate of MAA, and MAS.
Also, if the anode doesn't have any oxidizer deposit on it, then it takes quite a while for some to form.
But, if there is an oxidizer deposit already formed, then that deposit rapidly grows larger - towards the cathode, with the passage of more current.
I think I may have found a simple, working solution for making up the oxidizing paste.
Basically, a saturated cell (dark red/violet, like blackcurrent juice) is used to turn manganese-ammonium sulphate into MAA which immediately
precipitates out. My cell is saturated, any more MAA that forms, just precipitates out or forms on the anode.
Once the cell is made, enough MAS is added so that when enough current has been passed, you will have a saturated solution of MAA.
If too much MAS is added, you will have MAA precipitate, so it is not a problem.
With the saturated cell made up, start oxidizing kg quantities of MAS, that was prepared earlier.
Once it has been converted to the dark red salt, pour off as much of the acid/oxidizer mixture as possible. Remove the dark red salt and store in a
sealed acid-proof container until ready to use, then pour the cell acid (that you poured off) back into the cell.
Some of the acid will still be in the dark red "paste" you just removed, that is good since without it the MAA will decompose into MnO2 -
but after a few runs there will be no acid left in the cell!
That is fine if you only want a few kg of MAA, but if you want to make more more - you will need to replace that acid, and make sure the acid
concentration is unchanged. The occasional titration wouldn't do any harm to work out the acid concentration, over time it will be altered by
moisture, and water loss due to electrolysis. I'm reasonably confident this will work, because it's essentially the same process I've
been applying all along, only without the benzoic acid issues. But having to replace consumed acid, that's all.
So, you see - with this variation the saturated cell is just used as a "converter", converts MAS into MAA.
This way, no benzoic acid is ever formed!
The cell is NEVER heated! Before returning spent MAA (not MAS) back to the cell, make sure it is clean, and free of organics or you will end up with
the benzoic acid complications I encountered before. Avoid it. To clean up the MAS before regeneration, you can :
A) use low boiling solvents to clean up the mixture, then evorate at low temp.
or
B) Use steam distillation just for cleaning up the mix, but you will lose some of your sulphuric acid. The loss isn't that bad actually, but over
time you will have to keep replacing it.
I haven't yet tried my pH meter to work out the cell acid concentration, I would have thought that 60% H2SO4 is right off the scale...
Here is a photo of the MAA that I've been removing from the anode:
http://www.sciencemadness.org/scipics/oxidizingpaste.jpg
The next thing to do, is to work out a procedure for producing the MAS complex, without losses. This will involve mixing up stoichiometric proportions
of manganese sulphate and ammonium sulphate in sulphuric acid solution.
Again, the same solution (saturated) can be used over and over for making larger quantities of MAS, I'm sure. Otherwise, you will make up you
sulphuric acid, then precipitate your MAS (by adding MS to an excess of AS in sulphuric acid), and then what are you going to do with that acid
solution, throw it away? Make some diethyl sulphate? Who knows. It would be good to be able to use the same acid solution for making up ALL of your
MAS.
When I first made my cell, like I explained in an earlier post - I simply added MS to an excess of AS in the cell. My justification for doing this was
this :
"Well, if I make up the acid solution and collect the yellow MAS precipitate, I need to keep it wet so I still can't weigh it to work out
the exact quantity of oxidizer.."
But, if you use the acid solution, you may not know the exact quantity each time, but you can be pretty sure that it is reasonably pure. Again, and
again and again - without building up ammonium sulphate in your cell. If ammonium sulphate precipitates in the MAS acid solution, then it should be
obvious by it's colour.
However, I have 2 slight problems when it comes to scaling up:
1) I'm all out of MnSO4 reagent.
and
2) My transformer only puts out 2 amps.
I've been putting alot more than 120 amp hours to get a decent conversion. Probably due to organic acid that is still in the mix.
Needless to say, I won't be heating my cell to drive off solvent any more. Heating just cause more problems than it solves..
Believe me, I would only be too happy to make a 5 batch, 5 Kg, double-regenerating cell system, working on a rotating shift. Everything would be
recycled, and this process would run 24/7.
Heck, the more cells the better.
[Edited on 10-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Mass Production
For mass production of benzaldehyde from toluene, the air oxidation of toluene isn't so bad.
Probably alot easier for some than for others.
But if one wanted to go the electrochemical route, here is one way:
I should emphasise that this assumes recycling of the oxidizer. It is kinder to the environment to recycle the oxidizer, but not at all essential. It
could be skipped if one were in a hurry, simply by following the steps outlined in the last post to make a suitable quantity of MAA for a single
reaction - to make the desired quantity of benzaldehyde. That would shorten the following process greatly.
For 24/7 mass production, there are 9 main steps involved in this process, and some can be carried out at the same time (multi-tasking = good time
management!), and others can be combined - more on that later. Here they are:
1) MAA regeneration. May contain many individual cells, working in shift, with say 1 complete every 2 hours.
2) MAA settling. Pour cell mix into acid-proof container container, it must be left undisturbed for at least several hours to allow the MAA to settle
out at the bottom. This stage may be made up of a few containers, each holding one batch. When one is added for settling, one is transfered to the
next stage.
3) Pour off the saturated acid/MAA. This liquid will now go back to the cell for the next regeneration. You will now be left with a batch equivalent
of dark red paste, it will probably still have an inch of liquid on top, it will not interfere.
4) Put into storage container. It would be a good idea to make this container have a several "batch" capacity. So, lets just say that each
"step" involves 1Kg batches. Then build up a "buffer" inventory in this storage container of perhaps a few to several Kg of MAA.
That way, if an accident occurs or the power goes out, you can still process toluene 'round the clock; this inventory will act as a buffer for
your system - though not essential. Steps 1,2 and 3 simply add to this container, remove oxidizer in portions you are able to react at a time, maybe
3kg reactions at a time will be possible? Probably easier than 3 separate, time consuming 1kg reactions at 50 C.
5) Remove predetermined portion of MAA from storage container, and place in acid proof reaction vessel. This vessel must be fitted with some form of
overhead mechanical stirring. It should at lest have a partiall sealed top, to help contain odours (and minimise evaporation of your toluene). A large
excess of DRY TOLUENE is added. A large excess will be necessary if you are going to use mechanical stirring. This stirring mechanism would not be
hard to make, easy from a power drill and an acid resistant rod. A fast spinning axle with a small impeller should be good for really whipping up the
reaction mixture into a toluene/MAA milkshake. (The lower the temperature this reaction can be made to happen, then the better it probably will be).
6) Solvent (with large amount of dissolved benzaldehyde) is poured off. You may wish to add additional toluene to extract out as much of the
benzaldehyde as possible. Use the mechanical mixer for each extraction.
7) Washing the solvent. Wash with water to remove excess acid. You may wish to back extract this water to reclaim some residual benzaldehyde/solvent.
You could use a different solvent for the back extraction to conserve toluene. The water extracts could be fractionally distilled to recover some cell
acid.
***This benzaldehyde laden solvent is the output from this process, it is put into a storage container and is removed when practical to do so, for the
distillation process.***
8) MAS is transferred to solvent removal stage. This could be a vacuum vessel, or some kind of evaporator, or a solvent extraction using a low boiling
solvent. Preferably low boiling as in < 50 C low boiling, maybe a higher pb could be used, uncertain at this time. A combination of (low boiling
point) solvent extraction with subsequent vacuum evaporation could be used, or just plain old evaporation in a lowe humidity area, on a large acid
resistant sheet could be used? Lots of possibilities.
9) MAS goes straight back into saturated MAA cell solution (from step 3) for regeneration, in 1 or more separate cells.
***Distillation process is run under vacuum. The first distillation could be run at normal pressure to drive off the toluene, then what remains is
transferred to the vacuum distillation stage. Even a rather small vessel could do this, it would not have to be very big. I'm sure even a 250ml
vessel could handle the output from this process, unless your regen cells were pumping out one heck of a current...No need for large vac. vessels***
Okay, I bet you're thinking this all sounds kinda complicated, well - it doens't have to be. Some of the steps can not only run at the same
time, they can be combined.
For instance, 1 very large single cell could be run with a large hold up, say a few Kg solid material. Then, 1 Kg at a time can simply be removed when
needed for the reaction, and the expended oxidizer from the last reaction (MAS) put back into the cell when convenient to do so.
Using that approach solves alot of practical issues, it just means that the cell will never be 100% regenerated at any given time - but who cares?
Just keep an eye on the colour of the mix, keep it a dark-ish red, and make sure you keep organic solvent out of the regenerating mixture.
That large cell could also be the settling tank as well. Just switch it off for a while, it will settle pretty good in just a couple hours - if you
are in a hurry. A scoop can retrieve the red paste or sludge from teh bottom, it's not difficult, believe me.
For a large single cell, some kind of overhead stirring would be a good idea -though I'm unclear how essential it would be (at this time).
Chunks of oxidizer that collect on the anode could either be A) broken off and mixed EVENLY with the solid material or B) maybe collected and used as
the oxidizer. This would avoid the need to switch it off.
It is perhaps worth noting that this roto-batch style process - like most processes - has it's continual process equivalent, I think people oten
refer to that as "going technical"
The continual process equivalent would use a MAA saturated solution of cell acid, that is pumped via the process equivalent of all the above
steps/stages.
Only that instead of working with a solid mixture, you would be working with an acid solution (at some stages red, at others yellow), which is not
without it's own set of challenges...ie. acid-resistant pipes, pumps, level regulators, flow controllers, etc. would need to be obtained.
Or the batch system could be miniaturised to produce benzaldehyde more regularly, only on a more manageable scale.
Doing EVERYTHING in single individual stages is alot of work.
Although the above proposal may have a solid material (MAA and MAS) holdup of perhaps 8 or 10Kg, running 10kg batches in single steps would be a
nightmare, and very slow. It would be frustrating doing nothing, but waiting for your 10kg to regenerate.
If you were actually able to work with 10Kg batches, you would be thinking, hey I could be reacting another 10kg right now, and then extracting 10Kg,
and then washing...
Maybe someone will actually do that, there's an idea?
10 sounds a lot better than 1.
[Edited on 10-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Update
This is just an update on the reaction I started this evening.
Chemical used:
~430g Cinnamon aldehyde
120g sodium carbonate
2L boiled tap water

The 2 flasks at the front contain (from left to right) cinnamaldehyde and benzaldehyde. At the back: cinnamonaldehyde (green, freshly distilled),
undistilled toluene, and benzaldehyde from the last toluene oxidation run. Note the colour change of the cinnamaldehyde, it has changed from orange to
burgandy, since it was distilled 2 days ago.!

Fancy a cuppa' benzaldehyde anyone? Great! I'll just pop the kettle on!

The 120g sodiumcarbonate is still dissolving in the dish of hot water.
The magic ingredients. http://www.sciencemadness.org/scipics/2magicingredients2.jpg
Yes, thats right - I shop at Tesco supermarket, the best for quality low prices.
Not as high tech as my Heidolph MR 3003, but it will do the job.

Bumping granules. Pumice granules are generally much better for oily mixtures. I found a large lump of pumice in a field, and smashed it to powder
with a hammer.

(Glass left, pumice right)
Making up the oil bath.




A closer look at the oily mixture:
http://www.sciencemadness.org/scipics/2cyclooilydroplets.jpg
A dude's eye view of the overall setup:
http://www.sciencemadness.org/scipics/2cyclosetup.jpg
[Edited on 9-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Cinnamon experiment #2, update
I refluxed the mixture for 12 hours straight.
I started the reflux yesterday at 20:00,
and I switched off the hotplate at around 08:30 this morning. I came home from work at lunchtime and switched on the heating only for about 30 minutes
and after work I switched off the stirrer at 18:30.
Eclectic had suggested that I try steam distilling the mixture instead of using solvents to do the extraction directly.
This is what I'm doing just now.
There are many ways this could benefit the process.
If benzaldehyde distils off much faster than the residual cinnamaldehyde, then steam distilling will help to get the remaining cinnamaldehyde reacted.
At this time, this does APPEAR to be happening.
The magnetic stirring was a fairly subtle process, and I'm sure that any droplets that didn't get broken up overnight, sure are atomised
now.
This is the reaction mixture just before I started the steam distillation.

A closer look at the mixed top layer after the reaction, just before the steam distillation.
http://www.sciencemadness.org/scipics/2cyclotoplayermixed.jp...
This is the pot for the pressure cooker that I use to generate the steam.

This is an essential tool for steam distillation. It is just a bent glass tube connected to some rubber tubes, this makes it flexible and so
won't get damaged by the very turbulent mixture. The tube is bent 180 deg at the end to prevent rather disturbing back pressures that are
produced when a large bubble of steam is sudddenly released, this way the steam is always moving upwards through the mixture. This really helps to
maintain a constant flow of steam.
http://www.sciencemadness.org/scipics/2steamdistillationtool...
Here is the overall steam distillation setup. I'm sure it fairly self explanatory.
http://www.sciencemadness.org/scipics/2cyclosteamdistillatio...
Same again.
http://www.sciencemadness.org/scipics/2Cyclosteamdistillatio...
The steam is distilling through at a moderate rate.
http://www.sciencemadness.org/scipics/2cyclo3neck2.jpg
Oily droplets can be seen running down the inside of the 1L volumetric flask.

A closer look at the oil droplets.
http://www.sciencemadness.org/scipics/2cyclooildroplets.jpg
Here is the liquid that has been collected so far. This is the total amount of steam that has been condensed.

I guess the question is: Has the reaction been a success?
I could be real scientific (yawn..) and say that we will have to wait 'till the experiment is finished and the product distilled to work out the
composition.
I've tasted the steam condensate a few times, and I can say right now that the steam condensate tastes considerably "hotter" than the
pure cinnamaldehyde I started off with!
That must be a good sign.
There is clearly only a small percentage of total oil in the condensate.
Here are the latest images:
These pictures were taken just as the first litre had finished coming over, the oil at the bottom is approximately 50ml or more.
http://www.sciencemadness.org/scipics/cyclodrip.jpg
http://www.sciencemadness.org/scipics/sideview.jpg
http://www.sciencemadness.org/scipics/yield.jpg
http://www.sciencemadness.org/scipics/yield2.jpg
At one stage the cooling water stopped flowing because some of the cooling water boiled in the condenser.
When this occurred the benzaldehyde laden steam didn't condense and filled up the house with a seriously eye stinging cherry aroma, in just
several seconds.
I may have just found the world's most wonderful smelling tear gas.
It is now after 11pm where I live, and I will need to continue this tomorrow evening. So far exactly 2 litres have been condensed. It contains at
least 100ml of free benzaldehyde laden oil, though I think that is a conservative estimate - it may be as high as 200ml.
[Edited on 10-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Cinnamon experiment #2 results
Steam distillation has now been completed.
This is the mixture before I resumed the steam distillation this evening.

Rubber tubing dissolved, just used a straight glass tube for the remainder of the distillation. The tube on the left is the same time of tube before
being exposed to benzaldehyde, and the tube on the right it how it looks now. Benzaldehyde has turned the rubber tube in a cherry lollipop.
If I didn't know better, I would eat it.
http://www.sciencemadness.org/scipics/dissolvedrubber.jpg
Earlier I added approximately 3g of lecithin. As you can see, some foaming did occur. This was no problem, my vigreux made swift work of it.
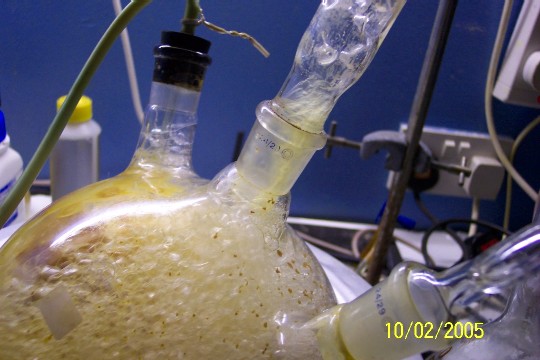
This is approximately 80% of the total oil product, not including the oil still mixed with the water. The condensate has yet to be extracted.

After the above photo was taken, about another 40ml of oil was recovered from further condensate.
In total, about 200ml of free oil has been recovered from the steam distillation. With each litre of condensate, there is about a tablespoon of more
product. So, here the point of diminishing returns is approaching..
I don't know how much benzaldehyde is still held up in the water, so far - I've condensed 6 litres (~1.5 gal.) of water, which has removed
most of the product. There is about another 200ml of oil unaccounted for.
I am quite certain that the free oil so far collected is almost pure benzaldehyde. If there is any cinnamaldehyde in it, it must be less than 5%. It
is not even slightly noticable.
If the other 200ml has been degraded and is still in the mix then it could be due to either the increased concentration, or it could be down to the
steam distillation - I am unsure.
I could try the reaction again, only shortening the reaction time to 4 hours or less, then start steam distilling. Perhaps a 12hr reaction + a day
stirring + steam distillation for 4 hours + cooled + steam distilled again the next day for another 4 hours is a bit too aggressive for the aldehyde
mixture. I will do it again, but I will need to wait until I get more cinnamon reagent.
Once I have extracted the 6L of water, I should have a better idea of the overall reaction efficiency.
I think this reaction is a good one to do while you wait for your cell to regenerate.
Speaking of which, now on to the toluene oxidation..
This is a picture of the cell + the pure oxidizer I have been removing from the anode.

Aren't they both so cute? The chemical equivalent of Dr. Evil and Mini Me.
They will both get put to work soon enough.
Note the colour, this is the colour of a fully regenerated cell. If I wanted to run a bigger reaction, all I would need to do is separate the
saturated MAA solution from the MAA, then add MAS and electrolyse and repeat until I had the total quantity I desired, topping up with acid solution
when necessary.
It just so happens I don't have the reagents to make up more MAS, at the moment at least.
Here is a recent shot of the anode, still encrusted with MAA oxidizer.

Note, that even with constant passage of over 2 A electrical current since this thread was started, there is still no discernible corrosion on either
electrode.
Only some discolouring is visible. These electrodes will last a very, very long time.
I may just keep my steam distillation apparatus assembled and use it to recover my benzaldehyde from the next toluene oxidation run, just like I used
to do. Having to replace some leached out acid now and again, isn't so bad really.
[Edited on 11-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Okay, this is what the mix looks like after the steam distillation.
http://www.sciencemadness.org/scipics/2finishedproduct.jpg

This is the total product, the free oil is on the left, the total DCM extract of the condensate is on the right.

This is the total extract from the condensate, after the DCM was boiled off. The oil weighs approximately 65 grams.

This is a photo of the benzaldehyde distillate, if you look closely you can see it was taken immediately as a drop had splashed into the benzaldehyde.
http://www.sciencemadness.org/scipics/2drophit.jpg
200g from previous experiment was added to the oil obtained from the steam distillation. Yield from this experiment is approximately 183g in very pure
form.
Final product, 383g pure benzaldehyde

The true total is probably closer to 400g, however I let some of the benzaldehyde distil over with the toluene so I would have pure benzaldehyde
without any toluene. I added about 50ml toluene to dry the benzaldehyde, hence the water-clear appearance.
183g from 430g isn't too bad. I did not plan to steam distil, it was a last minute decision.
Using a much shorter reflux time, and then steam distilling will no doubt produce much higher yields, IMO. I think it is definitely worth another try.
After I add in the benzaldehyde from my recent toluene oxidation experiments, I will have over 500ml of pure benzaldehyde.
Time well spent, I'd say.
[Edited on 11-2-2005 by CycloKnight]
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Not too shabby!
Well, there you have it folks - the end of chapter 1. Now go forth and be fruitful!
I would be very interested to know if anyone else manages to carry out either of these processes.
The more who do, the better. Purely for the benefit of the advancement of amateur science, that is. We amateurs have to look after ourselves ya'
know?
Please don’t hesitate to PM me if you have any questions I can help with.
I will carry out another scaled up variation of the cinnamon experiment at some point in the future, but it won't be for a little while yet. It
will probably be another steam distillation variation and then I’ll leave this process alone. It’s been done enough, by me at least.
In the mean time, I will be compiling another photo essay for a completely different process for a different thread. I will post it only when it has
been completed.
If all goes well, it should make very interesting viewing  . .
|
|
|
psychokitty
Harmless
Posts: 10
Registered: 5-12-2004
Member Is Offline
Mood: No Mood
|
|
You may want to read this before your last trial
( 1 of 1 )
--------------------------------------------------------------------------------
United States Patent 4,673,766
Buck , et al. June 16, 1987
--------------------------------------------------------------------------------
Method of producing benzaldehyde
Abstract
A method is disclosed for producing benzaldehyde by fractionally steam distilling benzaldehyde from cinnamaldehyde in the presence of hydroxide
catalyst and at a pH on the order of about 11 to about 13. Conversions of cinnamaldehyde to benzaldehyde can be achieved on the order of about 75% or
more.
--------------------------------------------------------------------------------
Inventors: Buck; Keith T. (Cincinnati, OH); Boeing; Anthony J. (Cincinnati, OH); Dolfini; Joseph E. (Cincinnati, OH)
Assignee: Mallinckrodt, Inc. (St. Louis, MO)
Appl. No.: 856595
Filed: April 25, 1986
Current U.S. Class: 568/433; 568/458
Intern'l Class: C07C 045/51
Field of Search: 568/433,458
--------------------------------------------------------------------------------
References Cited [Referenced By]
Other References
Guthrie et al., Can. Jour. Chem., vol. 62 (1984), 1441-1445.
Primary Examiner: Helfin; Bernard
Attorney, Agent or Firm: Wood, Herron & Evans
--------------------------------------------------------------------------------
Claims
--------------------------------------------------------------------------------
What is claimed is:
1. A method of making benzaldehyde com- prising
dispersing cinnamaldehyde in water,
converting the cinnamaldehyde to benzaldehyde under the action of heat in the presence of a catalytic amount of hydroxide ion and at a pH of about 11
to about 13,
fractionally steam distilling benzaldehyde and acetaldehyde from the cinnamaldehyde, and
recovering benzaldehyde from the distillate.
2. The method of claim 1 which is conducted at a pH in the range of about 12 to about 12.5.
3. The method of claim 1 wherein the benzaldehyde distillate resulting from the steam distillation is fractionally distilled for separation of the
benzaldehyde in substantially pure form.
4. The method of claim 1 wherein the acetaldehyde is vaporized during the course of the conversion while the benzaldehyde is condensed.
5. The method of claim 1 conducted in the presence of an anionic surfactant.
6. The method of claim 1 conducted under shearing agitation to facilitate the dispersion of the cinnamaldehyde in the water.
7. A method of making benzaldehyde com- prising
dispersing cinnamaldehyde in water in the presence of an anionic surfactant,
agitating the dispersion under the action of heat in the presence of a catalytic amount of hydroxide ion and at a pH of about 12 to about 12.5 for the
conversion of cinnamaldehyde to benzaldehyde,
fractionally steam distilling benzaldehyde and acetaldehyde from the cinnamaldehyde in a still having a pot temperature of about 105.degree. C. and a
column temperature of about 99.degree. C., and
fractionally distilling the benzaldehyde from the distillate for the separation of substantially pure benzaldehyde to obtain a yield of at least about
75% based upon the cinnamaldehyde.
8. The method of claim 7 wherein cassia oil is employed as a natural source for the cinnamaldehyde employed in the conversion.
--------------------------------------------------------------------------------
Description
--------------------------------------------------------------------------------
BACKGROUND OF THE INVENTION
The retroaldol reaction of cinnamaldehyde is well known. In this reaction, cinnamaldehyde is converted to benzaldehyde and acetaldehyde with various
potential side reactions. Recently, for example, an investigation of the kinetics of the retroaldol reaction of cinnamaldehyde has been reported by J.
Peter Guthrie, et al, Can. J. Chem., Vol. 62, pp. 1441-1445 (1984). While the conversion of the cinnamaldehyde to benzaldehyde has been long known and
well studied, it has not been heretofore known to produce benzaldehyde from cinnamaldehyde in substantial yields and favorable reaction conditions for
production of such yields have not been reported.
SUMMARY OF THE INVENTION
This invention is directed to a method of making benzaldehyde by conversion of cinnamaldehyde in the presence of water with surprisingly high yields
heretofore unachieved. The invention involves the dispersion of cinnamaldehyde in water and, in the presence of an effective catalytic amount of
hydroxide ion, fractionally steam distilling benzaldehyde from the cinnamaldehyde. The reaction is conducted at a pH on the order of about 11 to about
13 and, unexpectedly, within this pH range it has been discovered that a substantial conversion of cinnamaldehyde to benzaldehyde can be achieved on
the order of about 75% or more. It has also been found that the conversion may be achieved at such a high pH without adverse side reactions.
In a preferred mode of conducting the method, the cinnamaldehyde is dispersed in the water in the presence of shearing agitation and a surfactant. In
another aspect of this invention, it is preferred to employ an anionic surfactant such as sodium lauryl sulfate. Preferably, the hydroxide ion is
furnished by means of sodium hydroxide which also achieves the pH in the range of about 11 to about 13. It has critically been determined that the
fractional steam distillation of benzaldehyde from the cinnamaldehyde must be conducted at a pH within the range of about 11 to about 13, preferably
about 12 to about 12.5. Below and above this pH range, very poor conversions are obtained of 50% or far less and competing reactions interfere with
the production of benzaldehyde. Outside of this critical pH range, side reactions, polymerization and other adverse reactions prohibit any significant
yield of benzaldehyde. Yet, within the pH range of about 11 to about 13, especially about 12 to about 12.5, significant yields on the order of 75% or
greater are achieved and benzaldehyde is recoverable in substantially pure form free of side reaction products. These results are considered to be
unexpected especially at the high pHs of the reaction where it may have been expected that side reactions would have significantly lessened or
prevented the yield for the desired product.
During the course of the fractional steam distillation of benzaldehyde from the cinnamaldehyde, acetaldehyde is also vaporized and removed. The
removal of acetaldehyde thus prevents the forward polymerization reaction which otherwise competes in the presence of the catalyst. The benzaldehyde
which has been steam distilled is then subsequently fractionally distilled for separation of the benzaldehyde from other components in the distillate
such as minor amounts of acetaldehyde, terpenes and orthomethoxybenzaldehyde. It has also been found that a natural source for the cinnamaldehyde such
as cassia oil may be employed containing a substantial amount of the natural cinnamaldehyde. Thus, a natural product such as cassia oil may be
employed in the fractional steam distillation method of this invention and still the significant yields on the order of about 75% or more are
achieved.
DETAILED DESCRIPTION
The following detailed operating example illustrates the practice of the invention in its most preferred form, thereby enabling a person of ordinary
skill in the art to practice the invention. The principles of this invention, its operating parameters and other obvious modifications thereof will be
understood in view of the following detailed procedure.
OPERATING EXAMPLE
A solution was made up from 38.6 lbs. sodium hydroxide, 4 lbs. sodium lauryl sulfate and 10 liters antifoam agent in 760 gallons of water. The
solution was stirred until a homogeneous solution was obtained. Then, 1320 lbs. of cassia oil were placed in a 1150 gallon still. The oil contained
approximately 72% by weight of cinnamaldehyde. The still had a pot volume of about 1150 gallons onto which was mounted a 4 foot fractionating column
containing 1".times.1" ceramic tubes and a water cooled condenser was thereafter connected in series for condensing the benzaldehyde-water
azeotrope.
The above prepared sodium hydroxide solution was then added to the cassia oil and introduced into the pot of the still. The pot was equipped with a
stirrer. Using pressurized steam and vigorous stirring, the pot was heated to reflux with a pot temperature of 105.degree. C. Reflux was established
with a column head temperature of about 99.degree. C. Once reflux was established, it was continued for about 1 hour. During the course of the
conversion of the cinnamaldehyde in the cassia oil to benzaldehyde, pH was monitored and was maintained at about 12 to about 12.5. In the event the pH
fell below about 12, sodium hydroxide was added to bring the pH back up to the range of about 12-12.5. After refluxing for about 1 hour, take-off of
the water-benzaldehyde azeotrope was initiated. The water cooled condenser was operated at 100.degree. F. thereby enabling the water-benzaldehyde
azeotrope to be condensed and collected in a chilled receiver. The acetaldehyde by-product was principally vaporized at the temperature of the
condenser and was taken off as vapor. The distillate principally containing benzaldehyde in an amount of about 75% or more with minor amounts of
cinnamaldehyde, terpenes, orthomethoxybenzaldehyde and acetaldehyde was obtained. The crude benzaldehyde was thus collected in a chilled receiver and,
in a continuous feed operation the condensed water was continuously fed back to the still to replace what had been taken off and the distillation of
the azeotrope continued. The fractional steam distillation of the crude benzaldehyde continued until about 670 lbs. of crude benzaldehyde were
obtained. The crude distillate containing benzaldehyde was then dried under vacuum and fractionally distilled under vacuum of about 29" thereby
providing a boiling point for the benzaldehyde at about 70.degree. C. in order to obtain a substantially pure benzaldehyde free from residual terpenes
and other impurities.
Thus, by means of practicing the above process, the objectives of this invention are achieved in that cinnamaldehyde is converted into benzaldehyde in
substantially pure form even from the natural source of cassia oil. Surprisingly, it has been found that substantial yields in excess of 75% or more
of substantially pure benzaldehyde are achieved by this method. Moreover, it has been found that there is a surprising window of high pH at which the
conversion may take place in a fractional steam distillation column in order to separate the benzaldehyde and acetaldehyde from the reaction mixture
and still avoid the adverse side reactions from occurring.
Having described this invention and its operating parameters, variations may be achieved without departing from the spirit and scope hereof.
* * * * *
--------------------------------------------------------------------------------
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Another leap forward...
Very good work psychokitty!
I don't know how you found that, but that looks pretty darned good.
I'll carry out this new procedure for the last trial, with usual pics.
75% yields? Got to be worth a shot.
|
|
|
synthetika
Harmless
Posts: 1
Registered: 13-2-2005
Member Is Offline
Mood: No Mood
|
|
Toluene to Benzaldehyde
Hi all,
We too, are really interested in this particular subject,
We are now looking at some similar, and some new methods as well,
These pictures are just too good, They are sharp, and fruitfull with shit loads of needed, sometimes misunderstood info,
Syn
I believe that the persufate of Ammonia, works very well, having done it a few times, and the yields are very high
www.shroogle.org/synthetika
[Edited on 14-2-2005 by synthetika]
|
|
|
Taz
Harmless
Posts: 9
Registered: 5-3-2005
Member Is Offline
Mood: No Mood
|
|
cinnamon oil
Would it matter if the cinnamon oil is
derived from the bark or the leaves?
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
Yes
The leaf oil has a much lower concentration of cinnamaldehyde than the bark oil. You want the bark oil (or pure cinnamaldehyde if you can get it).
[Edited on 3-6-2005 by Polverone]
PGP Key and corresponding e-mail address
|
|
|
paccman278
Harmless
Posts: 13
Registered: 27-11-2004
Member Is Offline
Mood: do U care?
|
|
swip came across some mno2 today, how would one get to mnso4 from the mno2. would heatin it with sulfuric work? swip wants it for the electrocell.
|
|
|
solo
International Hazard
Posts: 3975
Registered: 9-12-2002
Location: Estados Unidos de La Republica Mexicana
Member Is Offline
Mood: ....getting old and drowning in a sea of knowledge
|
|
Re: Benzaldehyde from toluene
I recently ran into this threads about the current subject which may be of some assistance ........or not,....solo
http://home.ripway.com/2004-11/211899/toluene_benzylchloride...
http://home.ripway.com/2004-11/211899/everymans_toluene.txt
It's better to die on your feet, than live on your knees....Emiliano Zapata.
|
|
|
aldol
Harmless
Posts: 30
Registered: 7-3-2005
Member Is Offline
Mood: No Mood
|
|
benzaldahyde
the best way to benz is with bezyl alcohol
and reacting with mno2 works good
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Today I tried to make some benzaldehyde, just because it smells nice.
In reference to the method from neograviton I mixed 0,1mol MnO2 with 0,1mol conc. H2SO4, added water until it was a paste and evaporated the water
until white fumes appeared.
I added a solution of 10ml H2SO4 in 40ml water and stirred it for a while, it dissolved very slowly. After a while, I poured the black sludge into an
erlenmeyer flask and added 20ml toluene (reagent grade, extra pure) and stirred it rapidly with a magnetic stirrer.
After 30min. at about 50°C, there was not the slightest smell of benzaldehyde. I have no idea what I have done wrong.
The electrolytic method from CycloKnight is of course ideal for mass production, but the manufacture of the reagents consumes LOTS of H2SO4: first the
production of MnSO4, then the ammonium sulfate (I have to prepare this from H2SO4 and ammonia) and the cell again needs lots of H2SO4.
I considered setting up a small- scale electrolytic cell, but a cell containing 47,5g MAA, 45ml H2O and 79g 98% H2SO4 can, after electrolysis, oxidise
only 4g of toluene. Separating this very thin organic phase from the electrolyte would be very difficult.
Organikum posted another interesting process:
"300kg toluene and 700kg H2SO4 65% are mixed under stirring. 90kg MnO2 are added as fine powder under strong stirring. Temperature is kept at
about 40°C. After all MnO2 is added stirring is continued for some time, toluene and benzaldehyde is recovered by steamdistillation."
Maybe activated MnO2 (obtained by boiling in dilute HCl) is required. I'll try this out soon.
EDIT: Sorry, I didn't read the entire thread. The MnO2 route gives lousy results... hmm, seems like I have no other choice than to use the
elektrolytic method.
Didn't Polverone once speak of a catalytic process where toluene is oxidised with air?
[Edited on 1-4-2005 by garage chemist]
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
I am going to try out "activated" MnO2 in H2SO4 because nobody here tried this until now.
I boiled 20g of MnO2 (sold as "manganese black" from a pigment supplier) with 100ml 2,5% HCl for 1h 30min. There was no detectable chlorine
smell, but the chlorine might have dissolved in the very dilute HCl.
I tried to filter it and it totally clogged the filter, after 3 hours it still wasn't done. A large amount of MnO2 passed through the filter,
too.
I did a second batch and after boiling, I diluted it with 150ml water and I am letting it stand overnight so that the MnO2 can settle. Tomorrow
I'll decant it and let it dry.
I'll be away for a week (starting tomorrow evening) so I can't do any experiments.
|
|
|
aldol
Harmless
Posts: 30
Registered: 7-3-2005
Member Is Offline
Mood: No Mood
|
|
benzaldahyde
I no what you mean about hard to filter
you could try to let it settle out over night and then decant most of the filtrate first then filter.
or use a sintered glass funnell this will help heaps.
can anyone tell me why the sodium bisulphite addition is not contenplated
for the seperation of the aldahyde .
i no the yeilds are a little bit down but gee's it will make the seperation easier
|
|
|
transmuter
Harmless
Posts: 3
Registered: 17-4-2005
Location: gwondanaland
Member Is Offline
Mood: questioning
|
|
toluene_»benzaldehyde
Thanks Organikum.
Tried your "neograviton" posting & achieved a result. Only a small amount at this stage as I need to to make a small modification to
equipement. Will do another run within about a week looking for better results. Used MnO2 from a pottery shop, said it was tech grade. Cheap.
In regards the H2O2 method as per Pat. US 3531519, it makes very small quantities.The concern there seems to be the risk of creating to much benzoic
acid. Maybe a hybrid could be developed using H2O2 in place of Mn compounds. Pat. US 780404 & Pat. US 808095 both make the claim that under the
conditions perscribed no benzoic acid is formed.
Congratulations CycloKnight you have done a super job & thanks for making your progress available. May try the process at a later stage. One step
at a time.
Just a guess but maybe the buildup of your "anodemites" are due to the relatively low current being used as against what is recommended i.e.
2 amps instead of closer to 8.
|
|
|
Organikum
resurrected
Posts: 2337
Registered: 12-10-2002
Location: Europe
Member Is Offline
Mood: frustrated
|
|
| Quote: |
Garage Chemist wrote:
I mixed 0,1mol MnO2 with 0,1mol conc. H2SO4, added water until it was a paste
|
This cannot work when you add water and the method didn´t call for this.
The neogravitron method for sure works and it is the only non-electrolytic method known to me which does so with the MnO2 cement pigment of trade as
it is available to me.
I am working on reactivating the sulfate with H2O2 by now, as this is OTC here where I live (no good results up to now).
/ORG
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
So I have to mix the conc. H2SO4 with the MnO2 and heat it.
The problem is how to mix the two substances- I want to conduct the operation in a round bottom flask to minimize fuming from the H2SO4, but a
magnetic stirrer is not able to mix the MnO2 under the H2SO4.
Can I use an excess of H2SO4 to decrease the viscosity of the mix and facilitate mixing?
|
|
|
evil_lurker
National Hazard
Posts: 767
Registered: 12-3-2005
Location: United States of Elbonia
Member Is Offline
Mood: On the wagon again.
|
|
Swim is working on a new route to Benzaldehyde which is completely OTC from patent US 4,146,582.
Ferrous-Copper Catalyst: Toluene (7.6 g.), water (35 ml.), ferrous sulphate (0.110 g.) heptahydrate, cupric acetate (0.072 g.) and methanol (8 ml.)
are placed in a 250 ml. reactor.
Sodium persulphate (47.05 g.) in an aqueous-methanol solution of sodium persulphate is added slowly to the mixture which is maintained at 70.degree.
C., in an atmosphere of nitrogen and under agitation.
The organic phase is separated after two hours and the aqueous phase is extracted with ethyl ether.
The combined organic phases are distilled to afford 8.29 g. (95% yield) of very pure benzaldehyde (compared against a pure sample).
If this works it will be sooooo easy. The cupric acetate can be made by reacting a 1:2 molar ratio of dissolved copper sulfate (aka septic line root
killer) and sodium bicarbonate. The sodium bicarbonate is dissolved in about 2 liters of water and placed in a 5 gallon bucket. The CuSO4 is dissolved
in about 2-3 liters of water and then slooowly poured into the bucket. The stuff is allowed to sit over night while the Copper Carbonate Cu03
precipitates. The next day the stuff is washed with water by filling up the bucket and again allowed to sit. The water is decanted the day after and
again washed. After several washes, the Cu03 is then filtered out and a wad about the size of a pancake is placed onto a pizza pan sitting on a hot
stove eye. The heat breaks down the Cu03 into copper oxide, Cu0. The resulting finely ground Cu0 is then placed into a stainless steel pot of boiling
vinegar. The Cupric Acetate which is formed is thus decanted into a separate dish and then slowly evaporated.
The Iron Sulfate is prepared by again taking CuSO4 and dissolving it in water. Some unsoaped steel wool is placed in the vessel, and the CuS04 is
allowed to react. Copper precipitates as metal out of the solution and the iron sulfate is left as a green coloring in the water. The iron sulfate is
decanted off and dried similarly to the way the cupric acetate was.
The sodium persulfate is obtained at a pool supply store as a shock treatment. If the sodium persulfate is unavailable, it can possibly be substituted
with oxone or potassium persulfate and ajusted for weight.
To make the benzaldehyde, the ingredients are thrown in a pressure cooker large enough to hold many times the original formula. The pressure cooker
has been adapted in a manner to take out the pressure relief plumbing to fit it with a paint stirrer, and the safety relief is removed and equipped
with a rubber grommet and thermometer. While nitrogen atnosphere would be nice, it probably would not be necessary unless one's goal was to
minimize over oxidation of the toluene into benzoic acid.
After the reaction the contents of the pressure cooker are handled according to patent.
|
|
|
| Pages:
1
2
3
4
5
6
..
26 |
|












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}