| Pages:
1
2 |
blogfast25
International Hazard

Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
So, refluxing the EtOAc with say 50 % NaOH, followed by fractioning off the EtOH azeotrope? Sound like a route to fairly pure EtOH I've never
considered.
[Edited on 22-11-2014 by blogfast25]
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
Quote: Originally posted by blogfast25  | | So you reflux the ester straight over pure NaOH? No water whatsoever? If so, is it practical to distil off the EtOH while hydrolysis in on-going
|
Does not the thread title "ethyl acetate hydrolysis" and my own word "concentration" imply water...so I didn't put an "aq." before NaOH...
There's probably an ester hydrolysis in whichever lab manual; there happens to be one in Systematic Organic Chemistry that works.
|
|
|
Dronami_inc
Harmless
Posts: 13
Registered: 10-2-2011
Member Is Offline
Mood: No Mood
|
|
elegant solution 
If I understand properly, author used solid NaOH (not a solution)?
If so, obtained ethanol and acid are almost completely waterless. Please correct me if this is not so.
...I skiped this topic by accident and created a new thread https://www.sciencemadness.org/whisper/viewthread.php?tid=47958 where i'am interested in obtaining both anhydrous ethanol and
glacial acetic acid, but probably using gas phase hydrolysis with catalyst. How can it be implemented? i.e. without NaOH involved
|
|
|
Dronami_inc
Harmless
Posts: 13
Registered: 10-2-2011
Member Is Offline
Mood: No Mood
|
|
Thanks for all, the question A is still on the agenda
Bert, I took your suggestion and continued that branch
By the way, how do you think, what impurities other than water contained in technical EtAc?
|
|
|
Bert
|
Threads Merged
22-11-2014 at 16:18 |
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
It's probably not possible.
|
|
|
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
Neat and Cheap – EtOH, GAA!?
Okay, I'll probably appear to be cocky,this is not my intent, but could this thing be as perfect as it seems? I'm new at this stuff, so I need help. I
wanted to describe a summary of my intention. Please slap me around, where necessary.
First, a kind of Claisen condensation, of ethyl acetate with itself using a sodium hydroxide catalyst, is attempted. May as well go with a one molar
first run.
I'll set up for reflux, but have an ice bath ready, with one mole ethyl acetate( 98.23 mL) in a suitable(at least 2neck) RBF, say 500 mL. I'll use my
good, trustworthy reflux condenser and top it off with a calcium chloride guard tube.
I'll begin with moderate stirring and then add a small amount of my 40 g sodium hydroxide – POWDER, no aqueous solution. Observe the temperature of
the reaction mixture and add this mol of NaOH.
If I get 20 g sodium hydroxide in the stirring mole of EtAOc without appreciable temperature rise, I will hold here. I will look for signs of some
kind of reaction and perhaps the precipitation of sodium acetate prills.
If also, the thing begins refluxing exothermiclly, I will be happy, I will attempt to keep the reaction temperature under control(77 – 78°C) with
an ice bath. I believe that at this point the ethyl acetate will be beginning to be replaced by dry ethanol.
I will continue addition, until I've added the whole mole of sodium hydroxide. When the temperature begins to cool off, I may even heat this reaction
mixture. I can measure the pH, and odor (I told you, I'm new here).
Just judging things, I may continue refluxing. I'm not sure whether I need two equivalents of the ester, since I'm reacting it with itself. But it
seems like one acetate ion will be created by the condensation of each example of ethyl acetate.
The great thing about this is the possibility that we will be refluxing nearly completely ethanol at the endpoint and all of the sodium ions will be
hooked up with acetate ions. At this point I will treat the reaction mixture as hygroscopic, and switch over to regular atmospheric distillation
(still using a guard tube on the receiver. I think positive.) Theoretically I'll wind up with 58.4 mL dry ethanol.
Then (if by some a lark of a chance, or ultimately, after tuning) the still pot(having been very carefully distilled to dryness?) can be reset up with
an air condenser and an addition funnel with an appropriate amount of concentrated sulfuric acid and distill off glacial acetic acid!
Okay, now throw stuff. I'm going to try this, so please, offer cautions, considerations, modifications, etc. This would be a really cost justified
answer to the production of two elusive solvents which I need.
Some Notes:
1) even dry ethanol has some solubility for anhydrous sodium acetate. But surely, the atmospheric distillation with a double-sided, efficient
condenser should leave the solid material behind, right?
2) if the first reaction works and goes to completion,, then we won't have to worry about an ethyl acetate/ethanol azeotrope (EtOAc will be gone). I
don't expect much trouble separating excess concentrated sulfuric acid from GAA.
3) But, can we do the first reaction with no water? I think the use of an aqueous solution of sodium hydroxide leads to watery results, that are
difficult to get rid of. The idea of creating ethanol in an environment where water can't be formed is very intriguing.
4) It's damned convenient, about the boiling points of these two organic compounds. I mean, ethyl acetate boils at 77°C and ethanol just about 78°C.
It's terrible (impossible really) if you're trying to fractionate them apart. But of one is being converted to the other – it's just too convenient,
makes me suspicious.
[Edited on 26-12-2014 by CaptainPike]
|
|
|
Yugen
Harmless
Posts: 23
Registered: 20-12-2014
Member Is Offline
Mood: No Mood
|
|
This is too easy
What isn't already easy about making ethyl acetate? It's simple Fischer esterification. Aren't there already free resources for this synthesis everywhere? Check out page 73 of this free google ebook for example.
Is it the procurement of glacial acetic acid that bothers you?
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Uhm I am totally confused. It looks like captian pike wants to create ethanol and glacial acetic acid from ethyl acetate. I'm just entirely confused
why this post is here?
I don't know where to comment about the uhm... prep. Here or in the link?
|
|
|
Bert
|
Threads Merged
26-12-2014 at 09:25 |
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
THERE! I have officially gotten tomatoes thrown at me
Are you talking to me?
|
|
|
Bert
Super Administrator
Posts: 2821
Registered: 12-3-2004
Member Is Offline
Mood: " I think we are all going to die. I think that love is an illusion. We are flawed, my darling".
|
|
See your PM, not throwing tomatoes!
Rapopart’s Rules for critical commentary:
1. Attempt to re-express your target’s position so clearly, vividly and fairly that your target says: “Thanks, I wish I’d thought of putting it
that way.”
2. List any points of agreement (especially if they are not matters of general or widespread agreement).
3. Mention anything you have learned from your target.
4. Only then are you permitted to say so much as a word of rebuttal or criticism.
Anatol Rapoport was a Russian-born American mathematical psychologist (1911-2007).
|
|
|
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
Does anybody believe this reaction can occur (and go to completion) in a reaction media of ethyl acetate which changes to ethanol in the end?
CH3COOCH2CH3 + OH- ––––> CH3COO- + CH2CH3OH
I mean will the saponification continue with the last hydroxide ion attacking the last carboxyl? Or should I have an excess of either reagent?
I think it would be wonderful if the whole thing could occur with no water at all! But will it ever actually complete? And Yes, I am "of age", able
to buy booze, etc. etc. I just want to understand this reaction better. I also would like to see some glacial acetic acid (that I created) going in
and out of a solid form at somewhere around room temperature – don't you think that is cool?
And Another Thing – what if ethanol forms complexes with lye? And I mean, after the collapse of the tetrahedral, is there some way to protect the
newly formed ethanol from having its hydrogen(from the OH) abstracted or otherwise interfered with by the ongoing reaction?You can probably tell I
have very little experience in organic chemistry, either practical or hypothetical
PS: Happy New Year :-)
[Edited on 1-1-2015 by CaptainPike]
|
|
|
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
GAA/ethanol prep – Harmless Home Lab Version
Background
A one mole scale experiment was performed along the lines of a procedure posted by have2know, Acetic Acid Synth. Easy Home Version.
Have2Know's post outlined a two step process, first, the saponification of ethyl acetate by way of sodium hydroxide yielding sodium acetate and
ethanol and second, the pyrolysis (?) of the sodium acetate to make glacial acetic acid (either using sodiumbisulfate or concentrated sulfuric acid).
I intend to do both parts of the experiment, but only the first part has been so far attempted, and outlined here.
Some Initial Calculations and Assumptions
One mole of ethyl acetate, having density 0.897 and molar mass 88.11 grams/mol. 88.11 g of ethyl acetate will be 98.23 mL.
One mole of sodium hydroxide, in solid form 39.997 g/mol
one mole of ethanol having density 0.789 g/cm³ and molar mass 46.07 g would be 58.39 mL.
EQUATION: CH3COOCH2CH3 + OH- ––––> CH3COO- + CH2CH3OH [1]
MECHANISM: attack by a hydroxyl group – tetrahedral intermediate – kicked out alkoxide with rapid proton transfer creating the alcohol (resulting
in acetate ion) [2]
What I Did
In a nice, heavy, two necked, 250 mL round bottom flask was placed just under 40 g (one mol) sodium hydroxide in fine prills. This flask having one
major, central neck and the other splayed off to the side, was fixed just above a stirring hot plate, but initially in an ice bath. A thermometer was
placed in the off vertical neck, with the bulb extending below the midpoint of the flask but above the activity area of a stir bar which was then also
added. An efficient reflux condenser was fitted in the central, main neck and the circulation of ice water was begun. All tapered glass where joints
were greased with high temperature vacuum grease.
After a short time, roughly 99 mL (1 mol ) of hardware store ethyl acetate (Klean Strip MEK Substitute) was added in two separate approximately equal
additions. After a couple of minutes of heavy stirring, the 40 g of NaOH was suspended as a slurry in the one half mol of ethyl acetate. The
suspension remained at a temperature of 11°C with little or no exothermic action so the other portion of ethyl acetate was added, and a thermometer
adapter reinserted.
The ice bath was removed and the flask was heated gently on air bath. Shortly after reaching about 70°C, obvious refluxing began. This was allowed to
continue for approximately 2 hours after which time the liquid phase became limited and the solid volume increased markedly. Heating was increased
moderately in an attempt to melt the growing mass of crystals. Magnetic stirring was insufficient to move this mass significantly. Very little liquid
remained but this did continue to reflux.
The heating was stopped and a calcium chloride guard tube was added to the top of the reflux condenser to prevent the entry of moisture.
Observations And Discussion
One of the goals of this experiment was to produce ethanol and sodium acetate in an anhydrous form. Instead, some of the white solid material was
charred and darkened possibly due to oxidation. Very little liquid seems to exist separately from the wet solid.
I am at a loss as to what has happened. I could find no reference to any attempt at performing this saponification with no water added to the initial
liquid phase. My guess is that the reaction is halted somewhere short of completion. The still pot smells strongly, as one might expect, somewhere
between the ester and the alcohol. The crystals do appear damp and there is some liquid remaining. It was calculated that 58 mL of ethanol would
result from 100% conversion.
I attempted to put some litmus paper to a sample drawn out shortly after stopping. I'm not sure what it means the stuff is wet but not with water and
doesn't seem too weep into the paper. It may have represented significantly alkaline conditions.
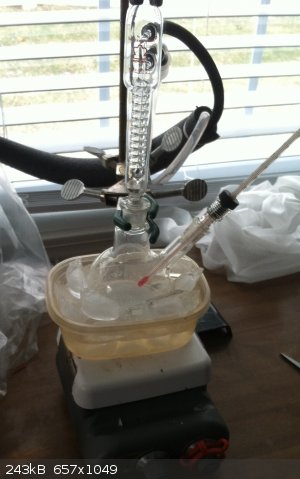
I have some photos. Something I've noticed since halting a couple of days ago. It seems as though the charred coloring has pervaded an ever enlarging
mass of crystals. Meaning, initially a dark spot was on the bottom, now the whole large mass of crystals has taken on the discoloration almost evenly.
A photo was taken shortly after shutting down using a cell phone. Then, I took some photos today with a somewhat better camera and a lower aspect.
Lighting, resolution, angle, camera and time are all changed, so surely my perception is subjective. But clearly the volume of white material is
larger now than when we halted a couple of days ago.
I also include a shot of my calcium chloride drying arrangement – I wanted to get something on there quick, before cooling down drew in appreciable
air. I wasn't able to pack my tube, plus, it's "college rule" (19/22). That adapter is a nice fritted bubbler (I had lying around), that fits
perfectly into that oddly shaped Erlenmeyer flask (stem almost touching the bottom). The barbed end of the fitted tube is unfortunately off screen,
but leads through a short piece of rubber tubing to a little 14/20 gas fitting in the top of the condenser.
And speaking of condensers, that little bantumware 14/20 reflux condenser is a really nice rig. It doesn't sweat but you can drive a lot vapor into it
and you won't even smell solvent in the room.
What to Do Next
I'm thinking I will rearrange the setup for normal distillation and pull off what liquid I can.
Alternately, I could add in some more dry ethanol , chop up the mixture with a stirring rod and try refluxing some more – push the
reaction further to the right?
Maybe this worked! I'm sure if there was pure ethanol, I have probably lost some through evaporation – could that really happen through the calcium
chloride packed tube? I could distill to dryness (using a heating mantle, instead of the hot plate?) and then continue with the pyrolysis using
concentrated sulfuric acid.
I could add in the slight excess of sodium pyro sulfate, begin the dry distillation, pulling off a for-run of whatever liquid is still present, and
then collect glacial acetic acid. (Well, it might happen!)
Questions
1) is this even possible – to do this reaction with no water? That is, starting with an ethyl acetate reaction media, then having this diluted with
the newly formed ethanol, ultimately ending in pure ethanol?
2) Relating to above, can I expect this theoretical reaction mechanism [2] to continue, unaffected by the increasing concentration of ethanol being
formed?
I would really appreciate any comments, suggestions, etc. I still haven't done anything else yet.
References
1. Chemical Engineering Education, Summer 2004, page 228
2. http://www.colby.edu/chemistry/PChem/lab/KineticsEster.pdf
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
This is a nice writeup, but why did you start a new thread? This is an identical topic and similar scale to the thread you mentioned.
|
|
|
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
Yes, I started a new thread.
Maybe as you imply, it should be just a post in the thread that I indeed mention.
It is basically the same thing done by have-two-know, but it's also different in that is done using glassware, etc. If the powers that be think it
should be moved in, as a post, or elsewhere, it is OKAY by me. I did have some discussion with O Most Powerful One, and I think it will stand alone. I
thought it might be better off over in the organic branch, even. So we shall see. Thank you.
|
|
|
Nicodem
|
Threads Merged
13-1-2015 at 12:54 |
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Dronami_inc | elegant solution
If I understand properly, author used solid NaOH (not a solution)?
If so, obtained ethanol and acid are almost completely waterless. Please correct me if this is not so.
...I skiped this topic by accident and created a new thread https://www.sciencemadness.org/whisper/viewthread.php?tid=47958 where i'am interested in obtaining both anhydrous ethanol and
glacial acetic acid, but probably using gas phase hydrolysis with catalyst. How can it be implemented? i.e. without NaOH involved
|
I got nowhere with this link. Is it dead, has it been moved or am I not allowed?
I'm trying to figure out whether this process can be done using pure lye, not a 50% solution or something. Once we start adding water ethanol doesn't
give it up so easily (nor does sodium acetate!)
I'd be interested in the opinion of S.C.Wack and others possibly following this discussion.
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
People always regret my opinionating. There is a way to do things and a way to isolate things and you're not there. You need lab manuals. This is why
I scanned several of them. Note the book that I suggested. And that there is a www button here. Perhaps djvu plugins are still available.
20 g of ethyl acetate are refluxed with 80 g (excess) of 25% aqueous caustic potash for 1 hour, until the layer of ester has disappeared, and the
mixture no longer smells of it. The whole is then distilled to 100°; ethyl alcohol can be separated from the distillate by addition of anhydrous
potassium carbonate. The residue in the flask is neutralised with dilute sulphuric acid and evaporated to dryness on a water bath. The solid residue
is powdered and distilled with 50 g of conc. sulphurlc acid to 130°, and the distillate fractionated between 115° and 120°.
Cooling in ice until solidification takes place, and subsequently draining away the still-liquid portion, gives crystals of glacial acetic acid.
Yield.-90% theoretical (12 g). Colourless liquid or crystals; characteristic odour; miscible with water; mp 16.7°; bp. 119°
|
|
|
CaptainPike
Hazard to Self
Posts: 68
Registered: 21-12-2012
Member Is Offline
Mood: No Mood
|
|
That wasn't too bad a, "Whack", after all!
Thank you, S.C., for the following procedure, which makes sense. I do have a handful of questions about what you have said which follows.
| Quote: Originally posted by S.C. Wack |
20 g of ethyl acetate are refluxed with 80 g (excess) of 25% aqueous caustic potash for 1 hour, until the layer of ester has disappeared, and the
mixture no longer smells of it. The whole is then distilled to 100°; ethyl alcohol can be separated from the distillate by addition of anhydrous
potassium carbonate. The residue in the flask is neutralised with dilute sulphuric acid and evaporated to dryness on a water bath. The solid residue
is powdered and distilled with 50 g of conc. sulphurlc acid to 130°, and the distillate fractionated between 115° and 120°.
Cooling in ice until solidification takes place, and subsequently draining away the still-liquid portion, gives crystals of glacial acetic acid.
Yield.-90% theoretical (12 g). Colourless liquid or crystals; characteristic odour; miscible with water; mp 16.7°; bp. 119°
|
Some Questions On the above:
1.) can I use sodium hydroxide ("caustic soda")?I have this on hand. Although it would seem that 80 g would be a lot of lye to use with 1/2 mol EtOAc
– straighten me out on this?
2.)What exactly is a 25% aqueous caustic potash? How do I prepare, "80 g of 25% aqueous"?
3.) "the whole is then distilled to 100°" – does that mean as opposed to dryness? The still pot temperature or the still head temperature (going
into the condenser)? You mean normal distillation and the temperature will rise and I stop at 100°C? Right?
4.) It seems like there will be more water (from that 25% aqueous solution of sodium or potassium hydroxide) in the distillate then can be absorbed by
a drying agent such as potassium carbonate. Oh, maybe that's why you say, "to 100°". My experience with distilling aqueous solutions of ethanol says
that if I take the fraction up to 100°, there will be appreciable water in the distillate.
5.)I always thought CaO was the best drying agent for ethanol. Is potassium carbonate better? What about sodium carbonate… and can I dehydrate that
to the anhydrous form by baking in a conventional oven to 525°F? I have some washing soda here on the farm. [Shucks]
– – – – – – – –
Help with any of these questions will be greatly appreciated.
I blundered on blindly and drew about 25 mL of some clear liquid that came over around 78° from my yellow colored sticky snow. I used an interesting
recombination of my apparatus and provide a crow's nest view of the set up. I have a lot to learn, but it sure is fun.
Attachment: phpsSVpCx (1.3MB)
This file has been downloaded 1089 times
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
The idea with the potassium carbonate is to form (at least) a saturated solution and separate layers, like NaCl and IPA. I've never tried sodium
carbonate. Hopefully you can figure out everything else.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
I tried this process a couple of times to obtain water-free EtOH over the past 2/3 days at a 2 mol scale.
In practical terms, the NaOH prills do swell up, presumably trapping any EtOH formed, as it becomes a solid lump.
Strong heating in a disty rig didn't even get close to any distillate after 10 minutes, and i suspect (not looked it up) expansion co-efficients would
end up with more cracked glass than product, so i stopped. (value the glass more than the booze).
When the solid mass was broken up there was still the distinctive smell of ethyl acetate.
The easiest way to get the lump out of your glassware is to add water, which kind of makes it useless for anhydrous EtOH production for an amateur.
Strong heating was noticed when the water was added, so presumably a lot of unreacted NaOH as well.
At a 0.25 mol scale or less, this reaction will probably work (for getting anhydrous ethanol) if stirred strongly for 24 hrs and with pure reagents.
For producing sodium acetate, it does work, although at bigger scales the yield of sodium acetate will be limited by how well you can thoroughly mix
slightly damp solids whilst controlling the temperature.
Personally i'll stick to vinegar/sodium bicarb and a lot of boiling, then a recrystallisation or two.
[Edited on 27-5-2016 by aga]
|
|
|
halogen
Hazard to Others
Posts: 372
Registered: 18-4-2004
Member Is Offline
Mood: No Mood
|
|
EtOAc + NaOH -> heat + other stuff
NaOAc + H2SO4 -> heat + other stuff
"there has to be an easier way!"
Maybe you could polymerize it in a hot tube to split off ethylene.
F. de Lalande and M. Prud'homme showed that a mixture of boric oxide and sodium chloride is decomposed in a stream of dry air or oxygen at a red heat
with the evolution of chlorine.
|
|
|
| Pages:
1
2 |