JAVA
Hazard to Self

Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
Procedure of tosylation of p-aminochlorobenzene
Hi guys,
I have performed the synthesis of p-aminochlorobenzene a couple of months ago. The problem is that this aromatic amine becomes yellow (and then red)
in a alkaline medium. I did store this amine in concentrated HCl and that was a clever idea.
My idea is to use sodiumbicarbonate or sodiumcarbonate to neutralize the HCl and extract the freebase in EtOAc several times.
After the purification process I would distilling off p-aminochlorobenzene under reduced pressure.
However, I think the last step isn't necessary since both p-aminochlorobenzene and TsCl are soluble in EtOAc. My question is: does this work ?
The reaction times of amine tosylation ranging from 1 hour to 16 hours (see references), is this a straightforward reaction that reacts spontaneously
under reflux ?
Also, some chemists say that the end product hydrolysed quickly with moisture in the air but according to a german chemist these tosylated amines are
fairly stable.
What about the storage of these compounds and how long can I store them ?
References about the reaction time:
Tosylation of amines
Procedure of the tosylation of aromatic amines
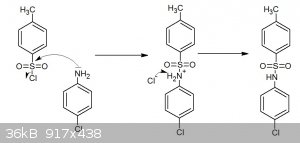
Java
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Java,
You want to run a very simple Schotten-Bauman reaction. It's described in most every sophomore college org chem lab manual. Read and learn! In a
nutshell. you can take your amine HCl solution, make it to about pH 13 with NaOH and shake the resulting mixture with a 10% molar excess of Tos-Cl. No
organic solvent is needed. Add additional NaOH during the shaking process to keep the pH above 11 or so. As the reaction proceeds, the tosyl amide
should dissolve in the aqueous layer. When the rection is complete (pH is stable and basic), acidify and filter off your tosylamide. Purify as you see
fit.
Your tosylamide should be stable indefinetely. Your particular tosyl amide will be extremely difficult to hydrolyze.
AvB
|
|
|
JAVA
Hazard to Self
Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by AvBaeyer  | you can take your amine HCl solution, make it to about pH 13 with NaOH and shake the resulting mixture with a 10% molar excess of Tos-Cl. No organic
solvent is needed.
AvB |
But tosyl chloride reacts with water (hydrolysis), I can read it everywhere. That's no problem ?
Secondly, p-aminochlorobenzene is a solid amine that is insoluble in water. Without organic solvent I should mix 2 solids and shake them. That doesn't
make sense.
The stability is yet another big problem since p-aminochlorobenzene becomes yellow if you don't use a inert atmosphere or a vacuum source.
I guess that nothing will happen except a rapid hydrolysis of tosyl chloride to the corresponding acid and a side reaction like aniline in air, but
way faster.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by JAVA | | But tosyl chloride reacts with water (hydrolysis), I can read it everywhere. That's no problem ? |
No, it is no problem, especially not with tosyl chloride. The Schotten-Baumann reaction is commonly used for the sulfonations of anilines or other
amines. I also used it for tosylation or anilines. That acid chlorides or anhydrides hydrolyse is more or less irrelevant by itself, what matters is
the difference in the nucleophilicity between the substrate and the chloride/anhydride.
The Schotten-Baumann synthesis of your product is in DOI: 10.1002/cber.19560890832.
| Quote: | | Secondly, p-aminochlorobenzene is a solid amine that is insoluble in water. Without organic solvent I should mix 2 solids and shake them. That doesn't
make sense. |
Both reactants have some significant solubility in water. I would personally first prefer using water with some THF, acetone, 2-propanol or
acetonitrile (4 : 1) and Na2CO3 as the base, but that's probably because I'm not too adventurous when I try a new reaction the
first time. Though, I think it might work exactly as AvBaeyer suggested it. Note also that under the Schotten-Baumann conditions, the yield depends on
the excess of the acid chloride and base.
| Quote: | | The stability is yet another big problem since p-aminochlorobenzene becomes yellow if you don't use a inert atmosphere or a vacuum source.
|
That is irrelevant. Coloration will not affect yields and has negligible effect on purity.
| Quote: | | I guess that nothing will happen except a rapid hydrolysis of tosyl chloride to the corresponding acid and a side reaction like aniline in air, but
way faster. |
The hydrolysis of tosyl chloride is notoriously slow and any reaction of aniline with air is negligible.
However, there is something I don't understand. If you are so concerned about doing it right the first time, why don't you used a published procedure?
What's wrong with the procedure in DOI: 10.1016/j.tet.2003.08.006 or DOI: 10.1039/C3CC40858B or any of the other reasonable examples? I think there
are even literature reports where an aniline and tosyl chloride are just mixed together, slowly heated up to release the HCl, and you are left with a
crude, perhaps just needing a recrystallization. Just don't do it on a large scale.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
I've done tosylation of 2-iodoaniline before using pyridine as solvent. It worked ok but purification was a pain. To re-attempt the synthesis now, I
would certainly use schotten baumann conditions. I've used that reaction to tosylate a primary alcohol (DCM solutions of substrate and TsCl, aq NaOH
with a little BnEt3NCl added as PTC), I've selectively N-acylated a 3-hydroxypyrrolidine with an aromatic acid chloride (THF/40% K2CO3 solution), and
several other successful reactions I don't remember well enough to cite.
The one time it did not work for me was in atttempting to N-acylate a phenylhydroxylamine with benzoyl chloride. This was later attributed to too fast
an addition rate and inadequate stirring, leading to a slight build up of HCl which effected the bamberger rearrangement to give the 4-aminophenol.
And as an old friend used to say; "colour doesn't weigh much"
[Edited on 24-10-2014 by DJF90]
|
|
|
JAVA
Hazard to Self
Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
I'll try it out, I don't suspect any problems actually. Stirring it just at r.t. for one hour is enough ? Heating might cause hydrolysis of the
product, right?
Final question is what solvent is well suited to remove the excess TsOH ?
Java
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
It should partition nicely into the aqueous phase. At end of reaction (check by TLC/NMR/any other suitable method) separate the phases and wash the
organics with a portion of water. Check pH is near neutral, otherwise wash again with water. Then just concentrate under reduced pressure to afford
your crude product (DCM dissolves very little water and forms an azeotrope). I'd suggest trying to recrystallise from (aq.) ethanol - this gave me
lovely colourless plates
|
|
|
JAVA
Hazard to Self
Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
Thank you DJF90 for the post reaction. In diluted hydrochloric acid the majority of the p-aminochlorobenzene was dissolved indeed. After adding aq.
sodium carbonate till pH 9 the free base was released as white powder while stirring at rt.
After addition of a slight excess of TsCl (pH 9) and stirring for 1 hour a brownish water insoluble chewing gum like compound did appear at the bottom
of the RB flask.
Now... the post-reaction 
Java
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Why didn't you use an organic solvent like I had suggested... likely would have avoided any gum, and provided a cleaner reaction.
|
|
|
JAVA
Hazard to Self
Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
It wasn't actually a sticky compound, before the post-reaction (still in the RB flask) it did look like a gum.
During the purification I isolated a crude brownish powder. I did dry that solid mass after washing it with water (3 times). The end product is very
soluble in pure EtOH and it looks a bit like whiskey at the moment.
I used a excess (220 mL) EtOH because I have time enough to wait untill the N-(4-chlorophenyl)-4-methylbenzenesulfonamide is visible as a crystalline
mass (th. mp. 120°C).
I did use 7,6 g p-aminochlorobenzene as limiting reagent.
No idea why the organic solvent is needed during the tosylation ?
I don't have a PTC to bring it into the DCM layer. Also, I didn't simply had any THF/Et2O during the experiment. Amines might react with acetone so I
didn't trust that and it's just a experiment.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Thats a fairly unconventional way of (re)crystallising your product. Personally, I'd dissolve in minimal hot ethanol, add water until a hazyness
persists, then add a little more ethanol to give a clear solution again. Then allow it to cool slowly to RT (or lower, if required).
Toluene is another very successful solvent for Schotten Baumann reactions. I'm not absolutely sure that the PTC is required, and have always added
some for good measure. It doesn't complicate the workup, and would do more good than harm.
|
|
|
JAVA
Hazard to Self
Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
I did already try to dissolve p-aminochlorobenzene in toluene, but the needle-shaped crystals quickly became yellow as well as the toluene.
Now I do understand what went wrong: p-aminochlorobenzene is soluble in diluted HCl but the freebase is a white solid and doesn't dissolve in the
aqueous phase. TsCl is soluble in water and might react faster with the hydroxide ions present. That is the reason why you did add THF: to dissolve
both TsCl and p-aminochlorobenzene so that they are able to react.
Unfortunately, after adding just a little bit cold EtOH (less then 20 mL) everything did dissolve, including the brown solid and suddenly a bloodred
solution was obtained.
Now I'm just curious whether I can remove the colored impurities from the end products. If there wasn't a tosylation then I'm still able to isolate
the unreacted p-aminochlorobenzene or if the "AvBaeyer procedure" did work I might end with the desired N-(4-chlorophenyl)-4-methylbenzenesulfonamide.
|
|
|
JAVA
Hazard to Self
Posts: 71
Registered: 9-1-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by DJF90 | | It should partition nicely into the aqueous phase. At end of reaction (check by TLC/NMR/any other suitable method) separate the phases and wash the
organics with a portion of water. Check pH is near neutral, otherwise wash again with water. Then just concentrate under reduced pressure to afford
your crude product (DCM dissolves very little water and forms an azeotrope). I'd suggest trying to recrystallise from (aq.) ethanol - this gave me
lovely colourless plates |
Do you have a IR spectrum of the lovely colourless plates, I see them on the wall and it looks like lovely colourless sodium chloride crystals.
I suppose the organics are still in the EtOH.
What might be the structure of the brownish coloured product?
btw:
All these full-text articles aren't free and you can't buy them without creditcard.
|
|
|








