MrHomeScientist
International Hazard

Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
Thermochromic Tetrachlorocuprate
For quite a while now I've been wanting to try making the compound smaerd shared in the Pretty Pictures (1) thread, and I finally have the reagents necessary. Here's progress so far.
Summary: Synthesis of lime green crystals resembling the desired product was successful, but they are highly unstable (tend to
melt, the copper in the compound is easily reduced to the metal) and I have not gotten them to change colors yet. They simply melt into a brown oil,
which returns to green but stays liquid upon cooling.
Procedure
Synthesis of thermochromic bis(diethylammonium) tetrachlorocuprate(II), ((CH3 CH2)
CH2NH2)2CuCl4 was attempted primarily following "Thermochromic Tetrachlorocuprate(II), An Advanced
Integrated Laboratory Experiment" by Choi & Larrabee (as well as smaerd's information). This is a bright lime green at room temperature, and
exhibits discontinuous thermochromism at 50C, changing to bright yellow.
Solution A: 2.20g diethylamine hydrochloride was dissolved in 15.1mL anhydrous ethanol with gentle heating and magnetic stirring.
Procedure uses isopropanol, but none was available and ethanol is listed as a substitute. This resulted in a clear solution.
Solution B: 1.70g anhydrous copper(II) chloride was dissolved in 3.2mL anhydrous ethanol with gentle heating and magnetic stirring. I
had quite some trouble getting this to dissolve completely, and after decanting from the solids only 1.60g had gone into solution. This was a very
dark olive green liquid.
Solution B was added to solution A, mixed completely, and transferred to a beaker while both were warm. The mixture was placed in the fridge to cool.
After cooling and waiting 1 day, the color had lightened to a "forest green" but no crystals were observed. Placed the flask on low heat to attempt to
reduce volume. Briefly accidentally heated to boiling before lowering the heat. Volume was reduced from ~22mL to ~15mL. Decanted this liquid into
another flask, and after cooling a bright green residue was left on the walls of the beaker. This exhibited the thermochromism on heating!
Unfortunately, though, nothing had crystallized in the rest of the solution even after cooling. Transferred 4mL of this liquid onto a watch glass to
try slow evaporation. This crystallized somewhat, but into dark olive green crusts instead of the lime green needles predicted by the procedure. I
left this out over the weekend.
Upon returning, all traces of the solids in both the beaker and watch glass had disappeared and I was left with a dark "oil." This oil was transferred
to a beaker and placed back in the fridge, after consulting with smaerd. After about a week, I returned to find the "oil" had completely solidified
into dark green crystals! Strangely, these liquefy rapidly upon removal from the fridge and warming up a bit.
I took a sample of these crystals and put them on a watch glass, placed under vacuum for ~40 minutes, to attempt to remove any remaining solvent. The
crystals remained solid while under vacuum at room temperature, but immediately upon releasing the vacuum most of them slumped back down to a puddle.
What I was left with, interestingly enough, was a small puddle of olive green liquid and lots of tiny lime green crystals. This liquid was drained off
as best as possible and returned to the cold beaker. Immediately, this froze back into the dark green crystals. The lime green
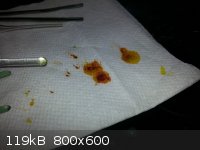
crystals remaining on the watch glass were transferred to a filter paper circle to absorb adhering liquid. The first photo is the crystals on the
filter paper, and the second is the remaining liquid on the watch glass.
 
These crystals appear to be stable, in that they do not melt back into a liquid at room temperature. (Why would these particular crystals be
any different?)
I took the very small amount of lime green crystals I recovered and placed them in a tiny vial, after adhering liquid had been absorbed by the paper.

I then took this vial and placed it on a pre-heated hot plate. Almost instantly, the crystals melted into a brown liquid drop. Removing from heat and
cooling back down, the drop changed to yellow-green, then back to a green color but still remained as a liquid. Severe overheating, perhaps?

IMPORTANT OBSERVATION: Apparently the crystals, especially when wet with the liquid, are highly sensitive to metal implements.
Scraping and transferring them with a metal spatula caused the oil to go almost totally black, which when wiped on a paper towel left rusty red
stains. This is almost certainly copper metal.
Finally, I have no information on how to dispose of this stuff. Since it looks like it's quite a fragile compound, I may be able to precipitate all
the copper by adding a suitable oxidant, say, hydrogen peroxide? But then what of the diethylamine hydrochloride leftovers?
This is apparently a difficult synthesis; more difficult than the publications would have you believe. I thought it would be a simple matter of
combining solutions and collecting crystals. All of my reagents are high quality, from Alfa Aesar, so the problems lie elsewhere. Interesting process,
though, that I'm enjoying exploring. I'll report back if I make any more progress.
|
|
|
TheChemiKid
Hazard to Others
Posts: 493
Registered: 5-8-2013
Location: ̿̿ ̿̿ ̿'̿'̵͇̿̿з=༼ ▀̿̿Ĺ̯̿̿▀̿ ̿ ༽
Member Is Offline
Mood: No Mood
|
|
Wow, this is great!
Are you going to upload a video of this to youtube?
When the police come
\( * O * )/ ̿̿ ̿̿ ̿'̿'̵͇̿̿з=༼ ▀̿̿Ĺ̯̿̿▀̿ ̿ ༽
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
I might if I ever get it to work! 
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Interesting compound, difficult synth, it would appear!
Destroy with strong NaOH solution and dispose of in general non-recyclable waste: these quantities are too small to worry about.
Good write-up, MrH S.
[Edited on 15-4-2014 by blogfast25]
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
Thanks blogfast.
I'm concerned with disposal because I plan to scale up if this works in small scale. I wantto ultimately make something suitable for stage
demonstration of the color change using a heat gun. What would the products be in the reaction with NaOH? I know next to nothing regarding organic
chemicals...
|
|
|
DraconicAcid
International Hazard
Posts: 4357
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
The reaction of sodium hydroxide with the diethylammonium cation will be diethylamine, which you could probably distill fairly readily, or react with
hydrochloric acid to get the diethylammonium chloride back. http://en.wikipedia.org/wiki/Diethylamine
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
The green crystals are exactly what you want. The yellow looks as though there is too much moisture. What % isopropanol did you start with before
trying to dry it with NaOH? Definitely try to distill the solvent before use if possible.
Also we crystallized from an ice/salt bath iirc. Which might be better then the fridge. Keep us posted!
edit - The red/orange color on your paper towel from my experiments on filter paper are a non-thermochromic byproduct likely from degradation
(moisture?).
[Edited on 15-4-2014 by smaerd]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
And the copper will be found back as Cu(OH)2.
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
Mostly I was looking for a way to neutralize the waste so that I would not need to set up a new waste stream for my lab (adds a lot of time and
paperwork). I'd filter off and discard the copper hydroxide, but I'm not sure what to do about the diethylamine. If there's no easy way to do this
I'll just make the waste stream, no big deal.
smaerd: On this run, I didn't use isopropanol. Anhydrous ethanol for both solutions. I'll distill my dried IPA and try another batch
with that, to follow the procedure exactly. I'm not sure where extra moisture would be coming from, since all my reagents except the diethylamine
hydrochloride are listed as anhydrous. If I remember right, you used hydrated copper(II) chloride and it seemed to work for you. My fridge is quite
cold - frost forms immediately on whatever you take out of it!
So you don't think the reddish stuff is metallic copper? Hm. Any other sources of info on this prep that you know of?
I placed some of the crystals wet with liquid in a dessicator, so we'll see what that does to them.
|
|
|
smaerd
International Hazard
Posts: 1262
Registered: 23-1-2010
Member Is Offline
Mood: hmm...
|
|
Okay one thing I had to do to dry the crystals was press them between several pieces of filter paper until the filter paper no longer came out moist.
I forgot about that. One of the intuitive processes you only think about in the lab I guess. Give that a shot on the green goopies.
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
I finally got a chance to distill my isopropanol, which was dried by shaking with 10% of its weight of NaOH then decanting from the saturated water
layer. I took the clear alcohol and distilled under reduced pressure in a rotovap. I had assumed that there would only be very little hydroxide
dissolved, and I'd only be removing traces of it. Turns out I was very wrong - and when about 80% had distilled over a crapload of NaOH dropped out!

I guess NaOH has decent solubility in isopropanol. This has got to be what fouled my second experiment using undistilled isopropanol. The distilled
product is crystal clear, and I'll give this experiment another go next week (hopefully).
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Alcohols do form alkoxides with bases like NaOH or KOH, although IPA isn't particularly good at this compared to methanol and ethanol. When I tried to
prepare potassium isopropoxide (KOH + IPA) some time ago, I found the KOH dissolved very well in hot IPA but on cooling most KOH dropped out again.
I'm no expert but for crude IPA, mixing with lots of salt gets out most of the residual water. Then distil. I think.
[Edited on 3-5-2014 by blogfast25]
|
|
|
Lambda-Eyde
National Hazard
Posts: 860
Registered: 20-11-2008
Location: Norway
Member Is Offline
Mood: Cleaved
|
|
There is a paper on the forum somewhere that discusses drying IPA, which calls for shaking with NaOH. It has been discussed many times before.
Interesting synth, btw. Another excuse to order some diethylamine. Good work!
This just in: 95,5 % of the world population lives outside the USA
Please drop by our IRC channel: #sciencemadness @ irc.efnet.org
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
Right, from what I read on the forum here saturating with salt is the first step to remove a good bit of water if you can only get the 80% variety of
alcohol. Then NaOH to pull out most of the rest, then distilling over a drying agent to finish it off. I didn't distill over anything, so there's
likely to be a small amount of water present but I'm hoping removal of the NaOH is what's going to do the trick.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by MrHomeScientist  | | Right, from what I read on the forum here saturating with salt is the first step to remove a good bit of water if you can only get the 80% variety of
alcohol. |
Aha. That explains why many, many moons ago when I distilled salted IPA the BP was so much off: water. Live and learn.
How anhydrous do you want yours to be?
|
|
|
MrHomeScientist
International Hazard
Posts: 1806
Registered: 24-10-2010
Location: Flerovium
Member Is Offline
Mood: No Mood
|
|
The paper I'm using specifies anhydrous solvents and reagents, but it looks like smaerd was able to make it with hydrated copper chloride so this
might be forgiving of some moisture. I can't think of a way to test the water content of my IPA, so I'll just try it as is and see how it goes.
By the way, here's the thread I referenced above. The drying method was posted by entropy51, in the green text.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by MrHomeScientist | The paper I'm using specifies anhydrous solvents and reagents, but it looks like smaerd was able to make it with hydrated copper chloride so this
might be forgiving of some moisture. I can't think of a way to test the water content of my IPA, so I'll just try it as is and see how it goes.
By the way, here's the thread I referenced above. The drying method was posted by entropy51, in the green text. |
Thanks MrHS, very useful info. I must have been distilling azeo IPA. (insert 'eyes raised to heaven' emoticon)
|
|
|









