| Pages:
1
..
28
29
30
31
32
..
45 |
DubaiAmateurRocketry
National Hazard

Posts: 841
Registered: 10-5-2013
Location: LA, CA, USA
Member Is Offline
Mood: In research
|
|
Alright,
So... what happened to the NO2 gas + Sodium acetate method ? I saw someone who tried it but did not report his result further.... on page 6-8
Also one person who used P2O5 seems to have got some acetic anhydride ? and that the crystal that form might be moist coming to the bottle that might
turned few Ac2O into AcOH and therefore it precipitated ? on page 4.
|
|
|
DoctorZET
Harmless
Posts: 42
Registered: 25-1-2014
Location: In the lab
Member Is Offline
Mood: tasting a pure sample of madness
|
|
Hi there, I'm new here!
I see you have big problems related to the production of acetic anhydride (at home).
That's OK, I can help you with some new methods.
(1)First method involves Sulfuryl chloride (O2SCl2) made by the reaction between SO2 and Cl2 in the presence of a powerfull light source (I used 500
Blue LED, but it work also with sunlight). After this you just have to mix carefully (with a glass stick) 2mols of sodium acetate (fine powder)
(anhydrous!!!) with 1mol of SO2Cl2(liquid at room temperature) to produce 2mols of CH3COCl (Acetyl chloride  ) and 1Mol of Na2SO4. In this reaction You can use a little bit excess of SO2Cl2. Then mix the Acetyl chloride(2 mols)
with another 2 mols of Na-Ac, and warm a little (45-49*C), to form the acetic anhidride (Ac-O-Ac ) and 1Mol of Na2SO4. In this reaction You can use a little bit excess of SO2Cl2. Then mix the Acetyl chloride(2 mols)
with another 2 mols of Na-Ac, and warm a little (45-49*C), to form the acetic anhidride (Ac-O-Ac  ) and NaCl. After this you can distilate an almost pure sample of acetic anhidride. Be careful, this reactions may
became hot enough to boil the SO2Cl2 or the Acetyl chloride, I recommend to add the reactants gradually. ) and NaCl. After this you can distilate an almost pure sample of acetic anhidride. Be careful, this reactions may
became hot enough to boil the SO2Cl2 or the Acetyl chloride, I recommend to add the reactants gradually.
[Edited on 25-1-2014 by DoctorZET]
|
|
|
TheChemiKid
Hazard to Others
Posts: 493
Registered: 5-8-2013
Location: ̿̿ ̿̿ ̿'̿'̵͇̿̿з=༼ ▀̿̿Ĺ̯̿̿▀̿ ̿ ༽
Member Is Offline
Mood: No Mood
|
|
Acetic anhydride in not Ac-Ac, it is Ac-O-Ac
When the police come
\( * O * )/ ̿̿ ̿̿ ̿'̿'̵͇̿̿з=༼ ▀̿̿Ĺ̯̿̿▀̿ ̿ ༽
|
|
|
DoctorZET
Harmless
Posts: 42
Registered: 25-1-2014
Location: In the lab
Member Is Offline
Mood: tasting a pure sample of madness
|
|
sorry about that...I mean Ac-o-Ac...However you may know that is impossible to make H3C-C(O)-C(O)-CH3 through these reactions...On the other hand
H3C-C(O)-O-C(O)-CH3 is formed. (I'm wrong if I consider that the acetate radical is "H3C-C(O)O-" instead of "H3C-C(O)-" ?  ) )
|
|
|
underground
National Hazard
Posts: 715
Registered: 10-10-2013
Location: Europe
Member Is Offline
|
|
Quote: Originally posted by DoctorZET  | Hi there, I'm new here!
I see you have big problems related to the production of acetic anhydride (at home).
That's OK, I can help you with some new methods.
(1)First method involves Sulfuryl chloride (O2SCl2) made by the reaction between SO2 and Cl2 in the presence of a powerfull light source (I used 500
Blue LED, but it work also with sunlight). After this you just have to mix carefully (with a glass stick) 2mols of sodium acetate (fine powder)
(anhydrous!!!) with 1mol of SO2Cl2(liquid at room temperature) to produce 2mols of CH3COCl (Acetyl chloride ) and 1Mol of Na2SO4. In this reaction You can use a little bit excess of SO2Cl2. Then mix the Acetyl chloride(2 mols)
with another 2 mols of Na-Ac, and warm a little (45-49*C), to form the acetic anhidride (Ac-O-Ac ) and NaCl. After this you can distilate an almost pure sample of acetic anhidride. Be careful, this reactions may
became hot enough to boil the SO2Cl2 or the Acetyl chloride, I recommend to add the reactants gradually.
[Edited on 25-1-2014 by DoctorZET] |
It sound interesting. Can you tell us more about the preparation of SO2Cl2 ?
|
|
|
Zwarts
Harmless
Posts: 2
Registered: 23-3-2014
Location: Europe
Member Is Offline
Mood: No Mood
|
|
Hello sciencemadness,
This is my first post here so please bear with me.
A good friend of mine works in stainless steel machine construction.
He is willing to make a setup for me in stainless steel 316.
This is how far i've gotten upto now.
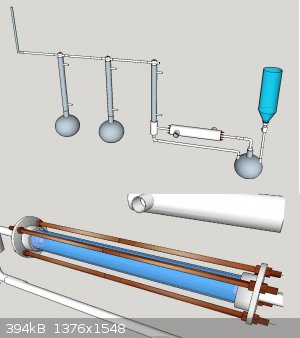
<img src="http://oi59.tinypic.com/inhcox.jpg" width="800"></img>
Basically acetone being heated.
Vapours lead through copper katalyst, cooled down through a ice water heat exchanger.
Condensed acetone recuperated.
Ketene led through gaa
ch4 recuperated/ disposed off
One of my main questions is what material i could use to connect the catalyst module to the rest of the piping
Also idd like you guys your opinion of it so far
[Edited on 3-4-2015 by Polverone]
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Have a look at this before committing yourself . . . ?
|
|
|
Zwarts
Harmless
Posts: 2
Registered: 23-3-2014
Location: Europe
Member Is Offline
Mood: No Mood
|
|
myea looks similar to me , tho the stainless steel setup would be a lot less fragile, glass, high temperatures, dangerous substances
considering this one would come at the same price nearly
|
|
|
Oxirane
Hazard to Self
Posts: 92
Registered: 19-9-2014
Member Is Offline
Mood: No Mood
|
|
Seems that only special part needed for that glass apparatus is the catalytic tube. It could be substituted with something common, though, perhaps.
I would try the design I attached. You can get the basic concept very easily. Few points:
The reactor is just a small tube, about 300-500mm in length from SS tube, diameter approx 20-30mm. It is surrounded by another tube, which has
diameter of appro 60mm inside, and this tube is insulated with ceramic wool or cast. Propane flame is blasted through the tube and is vented from the
other end as seen in the pic. It should bring the temp high enough for acetone pyrolysis.
It goes through this setup and vapors go into stainless steel adapter with silicon plugged condenser, which in this case contains as cold liquid as
possible, possibly insulated too. Unreacted acetone is condensed and led via PE/PTFE tubing back to the heating vessel to be revaporized and
pyrolysed. Generated ketene and other side products will go through the column and through another tube into second column. In here a second column
and flask is put if some acetone vapors shall pass, and one can use dropping funnel as well as presented in the document. The final flask contains GAA
into which ketene bubbles and hopefully reacts to produce AA. Rest of the fumes are led with a tube into atmosphere.
I think this same device could be used for making acetaldehyde, formaldehyde and ether by packing the reactor tube with proper silica gel supported
catalyst and using a digital hot air gun to control the heat, which is significantly lower in these reactions. In these cases, for formaldehyde you
could use very cool condenser coolrant, but in case of acetaldehyde, you would use coolrant at 20-30C and in case of ether, you would do it at 40C.
Also the first receiver flask would be put into controlled water bath to evaporate all ether/acetaldehyde but rest the ethanol down there.
There is also a close view of the reactor. It is presented in here as a quartz tube, but cheaper and much less fragile 316 will subside as well. The
ends are 1" stubs of SS tube filled with ceramic material and the flanges are steel.
[Edited on 7-11-2014 by Oxirane]
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
The major problem in this route seems to be the preparation of anhydrous sodium acetate, after that everything is "easy" (known)... The interesting
thing is the existence of sodium diacetate, which appears to be non-hygoroscopic, and in any case is generally prepared with water as a solvent:
http://www.scientific.net/AMM.508.79
A study of its crystal structure does not mention any hygoroscopicity or water of crystallization:
http://pubs.rsc.org/en/Content/ArticleLanding/1975/P2/p29750...
It appears the preparation of NaOAc*AcOH is much easier than the dehydration of the trihydrate of NaOAc.
Maybe you could use the dry EtOH/NaOEt solutions prepared here to neutralize and rextalize anhydrous NaOAc. I have a feeling that heating NaOAc*AcOH just gives
acetone and NaHCO3, though. If you're really lucky, NaOAc*AcOH might react with S2Br2 directly; it is dry, after all...
|
|
|
franklyn
International Hazard
Posts: 3026
Registered: 30-5-2006
Location: Da Big Apple
Member Is Offline
Mood: No Mood
|
|
If this hasn't been cited before it's worth a look.
NaHSO4 is heated to obtain Na2S2O7 and that then is heated with CH3COONa
http://chemistry.mdma.ch/hiveboard/novel/000462958.html
.
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
I'm afraid that reaction has been revisited dozens of times over the years, and it doesn't work at all. Similar reactions have been attempted with
P4O10, SO3, and NaPO3, among similar reagents (Al(OAc)2OH etc) which all fail to produce any anhydride.
I have however wondered if pyrosulfate could be used to dehydrate methanesulfonic acid to mesyl anhydride. While on the surface it sounds like it
would be even worse, sulfonic acids turn out to dehydrate more easily than carboxylic acids, and MsOH dehydrates to Ms2O with P4O10. Plus MsOH can
probably protonate S2O7(2-) which should make it more reactive. If even a little Ms2O is present at equilibrium you might be able to distill it off
with a little coaxing; the anhydride is significantly more volatile than the acid.
Or maybe it's all wishful thinking, but it would be pretty cool if it worked.
|
|
|
morganbw
National Hazard
Posts: 561
Registered: 23-11-2014
Member Is Offline
Mood: No Mood
|
|
I do not now and will maybe never, need the anhydride, but before I
would ever say that it clearly does not work I would have to design many experiments ( and test them to be sure ).
I have witnessed many things not possible for some but very doable for others. Just saying
To be sure, I have failed many synthesis.
[Edited on 1-3-2015 by morganbw]
|
|
|
franklyn
International Hazard
Posts: 3026
Registered: 30-5-2006
Location: Da Big Apple
Member Is Offline
Mood: No Mood
|
|
• Dry distillation of Calcium Acetate by itself leaves Calcium Carbonate and Acetone
. Ca(CH3COO)2 => CaCO3 + CH3COCH3
• Reacting Calcium Acetate with Sulfuric acid yields Calcium Sulfate and Acetic acid
. Ca(CH3COO)2 + H2SO4 => CaSO4 + 2 CH3COOH
• Reacting Calcium Acetate with the acid anhydride Sulfur Trioxide must make Acetic Anhydride. What else ?
. Ca(CH3COO)2 + SO3 => CaSO4 + (CH3CO)2O
• Skipping a step for a one pot synthesis :
. CaS2O7 + Ca(CH3COO)2 => 2 CaSO4 + (CH3CO)2O
Since Calcium Acetate discomposes at just 160 ºC, and Calcium PyroSulfate not until 720 ºC
that presents a problem. However Sodium Acetate melts at 324 °C boils at 882 ºC without
decomposition. Sodium PyroSulfate melts at 401 °C boils at 460 ºC without decomposition.
As it is detailed here _
Attachment: Acetic Anhydride Manuf US1430304.pdf (193kB)
This file has been downloaded 888 times
http://books.google.com/books?id=yZ786vEild0C&lpg=PA161&...
SO3
http://www.sciencemadness.org/talk/viewthread.php?tid=5495
The Thermal Decomposition of Potassium and Sodium Pyrosulfate
http://doc.utwente.nl/68103/1/Vries69thermal.pdf
Closely related, previous post
http://www.sciencemadness.org/talk/viewthread.php?tid=9&...
.
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
Energy, people.
CaSO4: -1434 kJ/mol
Ca(OAc)2: -1479 kJ/mol
SO3: -395.7 kJ/mol
Ac2O: -625 kJ/mol
-1479 + -395 > -625 + -1434
I think we might in fact have ourselves a winner. Unfortunately, this route is even worse than the last one... :p
|
|
|
franklyn
International Hazard
Posts: 3026
Registered: 30-5-2006
Location: Da Big Apple
Member Is Offline
Mood: No Mood
|
|
Am I the only one with imagination here. Mind you I'm not the chemist so how am I the one that conjures these
things.
Pyrolyze Potassium Bisulfate to produce Potassium PyroSulfate
2 KHSO4 => H2O + K2S2O7
Potassium Bisulfate decomposes at ~ 214 ºC
www.ilo.org/dyn/icsc/showcard.display?p_card_id=1585
Heat Potassium PyroSulfate and Sodium Acetate to distill Acetic Anhydride
K2S2O7+ 2 NaCH3COO => K2SO4 + Na2SO4 + (CH3CO)2O
Initially the precursors will melt and Acetic Anhydride will boil out leaving dry solid Sulfates. What can go wrong ?
Sodium Acetate NaCH3COO melts at 324 °C
Potassium PyroSulfate K2S2O7 melts at 325 °C
www.caslab.com/Potassium_pyrosulfate_CAS_7790-62-7
Sodium Sulfate Na2SO4 melts at 884 °C
Potassium Sulfate K2SO4 melts at 1069 °C
.
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
| Quote: | | What can go wrong ? |
You know, I'm really glad you asked, because if nobody had asked I wouldn't have thought about it anymore. Anywho, the definition of insanity is to
try the same thing over and over expecting different results. And now, may I introduce you to Mr. Clay S. Encondensation.
Step 1 is the evolution of SO3. It might be P2O5 in some other synthesis, but, like, who cares? We start with SO3; that one's easiest.
NaOAc + SO3 >> NaO(SO2)OAc, "sodium acetyl sulfate" -- a known compound (though to be fair I don't actually know if it forms in this
reaction -- I was just thinking about possible pathways that the combination would follow)
Anywho, sodium acetyl sulfate has an active methylene on the "acetyl", and since NaOAc is capable of deprotonating Ac2O you bet it's capable of deprotonating acetyl sulfate. On the other side, acetyl sulfate is itself a pretty
powerful acylating agent. Thus:
2 NaO(SO2)OAc >> NaSO4H + NaO(SO2)OAcAc
NaO(SO2)OAcAc >> CO2 + Me2CO + NaSO4H
A drastically more energetically favorable reaction than the production of Ac2O, and we already know that acylative decarboxylation occurs by heating
some acetate salts all by their lonesome. I figure the self-condensation of acetyl sulfate (and probably acetyl phosphate) explains all the mess --
and possibly the overheating, charring, etc...
[Edited on 4-3-2015 by clearly_not_atara]
[Edited on 4-3-2015 by clearly_not_atara]
|
|
|
learningChem
Hazard to Others
Posts: 182
Registered: 21-7-2011
Member Is Offline
Mood: No Mood
|
|
So, if there's any reaction between sodium acetate and sodium pyrosulfate, it doesn't yield acetic anhydride anyway?
If that's the case, then I've got a couple of questions :
Why do people bother to fill fake patents like US 1430304?
What kind of dumb joke is a patent system that allows outright lies to be 'patented'?
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
What does it matter if the patented thing is useless to the patent holder and they paid the fee? Patents aren't meant to be a how-to guide for third
parties, but a legal protection for an inventor so they can (for a limited time) capitalize on their work without being immediately ripped off by
competition. In order to have that legal protection they have to elaborate on what they invented so they can point a finger in court when another
corporation/entity starts doing it.
If the patent is garbage, it is worthless to the inventor. In order to keep garbage patents out of the system, the patent office would have to have
experts on everything on staff and test every single patent.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Okay, so the Na acetate + pyrosulfate method doesnt work, right? But the sulfuryl chloride + sodium acetate works. Or maybe it needs some catalyst?
And what is the mechanism of formation of acetyl chloride via sulfuryl chloride? For some reason there's almost no references for this reacton in the
modern literature: there's one patent that leads to monochloracetic acid, and there's nice article in the thorpe dictionary of applied chemistry about
acetic anhydride using SO2+Cl2+NaOAc in glacial acetic acid (SO2 forms adduct with acetic acid).
And does anyone finally found the mechanism of acetic anhydride(AA) formation from NaOAc+S+Cl2/Br2? Because the Ac-O-S-S-O-Ac most likely is a
bullshit. And we know that the reactian actually does not starts easily for some reasons, so there's no scientific research on this mechanism. But
sodium benzoate + S + Cl2 readily gives benzoic anhydride. I'd say sodium benzoate would be a nice catalyst for the reaction, though I never tested
it.
I think the actually catalyst for the old industrial preparation (NaOAc+S+Cl2) was a carbon obtained from fusing of the sodium acetate. But what
happens in the process? Stochimetry of the yield says that 2/3 of it can be made via sulfur oxidation product SO2 by converting it into sulfuryl
chloride SO2Cl2 and then reacting with sodium acetate as usual. But 1/3 of the yield is a mistery. Can it be a thionyl chloride? Maybe some
Ac-O-(S=O)-O-Ac intermediate.
We know that after the reaction finished, on heating to AA boiling point (140°C) the SO2 gas comes off. Also, we know that sulfuryl chloride
decomposes to SO2 and Cl2 above 100°C. Also, we know evolution of SO2 during the AA distillation from the final mixture leads to lower yields (Thorpe
says so), this is why vacuum distillation is used.
And did anyone try the Al2(SO3) + NaOAc method?
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
I guess it makes a little more sense to assume that the adduct between S2Cl2 and NaOAc has a form like a thio-thionyl dihalide, or S=S(OAc)2,
producing an Ac-O-S(=S)-O-Ac. Maybe AcOSSCl rearranges to AcOS(=S)Cl, which looks vaguely reasonable. Or even AcSS(=O)Cl, which is lower energy -- and
the resulting hemithio-dianhydride AcSS(=O)OAc has a sort of reasonable degradation pathway.
In any case the degradation byproduct is a known compound, providing a rationale for the process:
http://en.wikipedia.org/wiki/Disulfur_monoxide
I think the real question is whether someone can find a reasonable way to produce pure S2Br2. All of this in situ bullshit hurts yields and
makes everything smell bad.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
You don't need the S2Br2, you need a SBr2 if you want to make anhydride.
What would drive the reaction towards yielding Ac-O-S(=S)-O-Ac ?
I've found some answers in the article "Dialkoxy disulfides and their branch-bonded thionosulfite isomers"
DOI: 10.1080/1741599342000202176
"12a–12c synthesized by Wang [46] in excellent yield (>90%) are included for completeness (ROAg + S2Cl2, under vacuum). These compounds are
thermally unstable and decompose readily to form the anhydride, SO2 and sulfur [47].
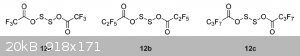
[46] Wang, C. S., Pullen, K. E. and Shreeve, J. N. M., 1970, Inorg. Chem., 9, 90.
[47] Denham,W. S. and Woodhouse, H., 1913, J. Chem. Soc., 1861."
They say the -O-S-S-O- isomer is the most stable one.
And here's a pic from that doc, though it describes slightly different compounds.
I was unable to find the 46 reference ( Wang, C. S., Pullen, K. E. and Shreeve, J. N. M., 1970, Inorg. Chem., 9, 90 ), but the 47 is available - https://www.thevespiary.org/rhodium/Rhodium/Vespiary/talk/fi...
And also this, written by one of the authors of the previous article https://www.thevespiary.org/rhodium/Rhodium/Vespiary/talk/fi...
Although those are way too old.
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
SBr2 doesn't exist! I'd be surprised if it were an intermediate, and no source (including yours) has suggested this. Besides, the reaction obviously
works with S2Cl2, and apparently by simple scission. The mechanism isn't really hard to understand -- what people forget is that sulfur has a really
large covalent radius, which allows sulfur compounds to undergo reactions that ordinary compounds don't. For instance, cyclizations of sulfur
compounds can violate Baldwin's rules:
http://pubs.rsc.org/en/Content/ArticleLanding/1976/C3/c39760...
In this case one of the acetyl groups attacks the oxygen on the other one, and gives off S2O, or at least that's my guess. The analogous reaction with
SBr2 seems if anything less favorable.
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
Truly, my sources still don't answer the question about maximum yield stochiometry of
8NaOAc + SCl2 + 2Cl2 = 4AcOAc + 6NaCl + Na2SO4
which is based on practical observations (thorphe 1921, vol. 1, p.28)
As far as I understand, the SBr2 is relatively unstable and is gaseous - the latter one is a more important problem. Just like SOBr2 is unstable
compared to SOCl2, and SCl4 is unstable. I'd suggest using chlorine instead of bromine.
Speaking about sulfur large size, I can mention sulfur mustard and willgerodt-kindler reaction with 3-membered rings.
Nevertheless, disulfur dichloride is not reactive enough to be successfully attacked by carboxylate, and, as you could see, researchers were forced to
use a silver salt, because they failed with sodium. I thought the mechsnism of NaOAc+SO2Cl2 reaction would help me to answer the question, though I
still don't have the mechanism.
|
|
|
clearly_not_atara
International Hazard
Posts: 2835
Registered: 3-11-2013
Member Is Online
Mood: Big
|
|
There is one serious problem with your hypothesis: silver makes carboxylate salts less nucleophilic, not more nucleophilic, which is
why silver acetate reacts with bromine: Br+ won't react with AcO- (even in solution), but silver's electronegativity of 1.93 (compare Na 0.93) means
that the Ag-O bond has covalent character whereas the Na-O bond is decidedly ionic (oxygen's electronegativity is 3.44; the rule of thumb for ionic
bonding is a difference of 2, which is seen by considering how AgF is soluble but Ag2O is not). Instead silver allows the salt to undergo other complex sorts of reactions, which is why the Hunsdiecker reaction occurs in nonpolar solvents where AgOAc reacts with
molecular Br2. Another example is the Kolbe reaction in ether which produces isonitriles because the carbon atom is essentially covalently bonded to Ag and so the nitrogen is alkylated
instead.
This might be why diacyloxydisulfides are not formed:
S2Cl2 + NaOAc >> NaCl + AcOSSCl
AcOSSCl + NaOAc >> Ac2O + S2O + NaCl
where in the second reaction AcOSSCl behaves as an acyl pseudohalide and acylates AcO-, and the electron in [O-]SSCl is transferred to chlorine to
leave S2O. Using silver salts prevents acetate from being acylated directly, possibly due to steric hindrance, or due to silver's (soft acid)
affinity for Cl- (soft-ish base) outcompeting AcOSSCl's affinity for acetate.
Sulfur-halogen bonds are very weak in general. It would be surprising if any of them don't add to acetate; they add to, for example, benzene in the presence of AlCl3, or with aniline uncatalyzed, and we can reasonably assume AcO- reacts about 60 times slower than aniline (which reacts essentially instantaneously, and
not only that, it reacts twice!. You'll notice that on Rhodium's Archive the yield is more than 90% based on S2Cl2, which makes a mechanism dependent on oxidation seem pretty unlikely.
[Edited on 24-4-2015 by clearly_not_atara]
|
|
|
| Pages:
1
..
28
29
30
31
32
..
45 |








