| Pages:
1
2 |
lilly
Harmless

Posts: 1
Registered: 29-8-2009
Member Is Offline
Mood: No Mood
|
|
cinnamic acid
The mild oxidation of cinnamaldehyde should obviously yeild
cinnamic acid. I have read of no one doing this.I think a rhodium file I read said permanganate on a solid support oxidized a cinnamic acid to the
benzaldehyde.
Can anyone suggest a procedure with a good chance of
oxidizing to the acid with out touching the double bond.
i have Mno2 and peroxide any ideas as to best solvents and reagents to try would be most helpful.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
All you need is an oxidant that does not cleave or alter double bonds. MnO2 is no good as it is designed to oxidise allylic alcohols without
overoxidation to the acid (i.e. it stops at the aldehyde). Perhaps jones reagent is applicable here, although id double check that... Sodium chlorite
(NaClO2) is also used specifically for this kind of transformation (i.e. aldehyde to acid, in the presence of a,b-unsaturation amoungst other things).
Workup would be much easier and cleaner than using jones reagent/chromic acid.
In fact, conversion of allylic alcohol to allylic acid is preferably done in two stages using MnO2, then NaClO2, rather than the "simpler oxidation"
with chromic acid because it is generally cleaner and higher yielding. Obviously you wouldnt need the MnO2 oxidation as you're starting with the
aldehyde.
[Edited on 29-8-2009 by DJF90]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I guess you could use H2O2 with a iron catalyst, have a look in the "oxidation of benzylic alcohols" thread for details, I don't think you will have
epoxidation in none-basic envinronement. Hey even NaOCl could do the job, if buffered.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Yes H2O2 oxidation in a non-basic environment may also work, but dont adjust the pH too low or you will have significant decompostion of the peroxide.
I don't even think an iron catalyst would be needed Klute, but I havent seen the thread so I'm not in a great position to comment on that. There are
numerous reagents that will work for the reaction, its just a matter of finding the one that works best, and often one method will excel far above the
others.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
cinnamic acid via the Perkin reaction
I have recently prepared cinnamic acid via the Perkin reaction per Brewster et al, 1960. This a base (NaOAc) catalyzed condensation reaction between
benzaldehyde and Ac2O.
In an 8" x 1" test tube 10.1 mL of benzaldehyde (homemade), 11 mL Ac2O (homemade), 1 mL pyridine (homemade) a catalyst, and 12g of technical anhydrous
NaOAc were mixed with a stirring rod. The NaOAc did not dissolve well, most remaining as a slurry.
The reaction tube was equipped with an air condenser and placed in a hot oil bath (silicone brake fluid, DOT 5). The bath was placed on a stirrer
hotplate. The hotplate was powered through a PID controller with a thermocouple for temperature control. The heating schedule was: 2 hours at 145C
followed by 5 hours at 175C. Some tar formed right away and turned the reaction mix dark brown. Also, much of the NaOAc settled to the bottom of the
tube. See photo below:

Temperature control was quite satisfactory throughout. When the gummy reaction mix had cooled it was laboriously removed from the test tube via
spatula and placed in a 1000 ml RBF with 250 mL distilled water. The tarry mix was then steam distilled to remove unreacted benzaldehyde. This
produced about 125mL of condensate. The product was then basified with NaOH solution. This was then filtered into a 1000mL Erlenmeyer flask:
[Edited on 4-9-2009 by Magpie]
[Edited on 4-9-2009 by Magpie]

The filtrate was then acidified by addition of muriatic acid. This resulted in the formation of a massive amount of fine flake crystals of cinnamic
acid. This was Buchner filtered and the cake redissolved in 850 mL of boiling water. The flask was allowed to sit for a few hours while the cinnamic
acid recrystalized. The crystals were again Buchner filtered then allowed to dry overnight.
The 3.7g of crystals were very light and fluffy, and had a slight yellow cast, which I suspect is from residual tar. Melting point was 134C which is
in agreement with literature values for trans-cinnamic acid. Expected yield was 61%, actual yield was 25%, based on the amount of benzaldehyde
charged.
[Edited on 4-9-2009 by Magpie]

Discussion
Although Brewster mentioned that there would possibly be some tar I was very surprised at the amount. Perhaps my homemade, somewhat impure, reactants
contributed to this.
All the undissolved NaOAc was also somewhat plagueing. This residual just formed a hard mass on the bottom of the tube. I doubt the procedure
authors intended that result.
Next time I would pour the reaction mix into the RBF before it had a chance to cool and solidify.
When I saw the heavy tar formation early in the heating period I didn't have much hope for any yield. I am happy with the product as the mp indicates
high purity despite its yellow cast.
I'm looking forward to making some hopefully nice smelling esters with the product using methyl, ethyl, and benzyl alcohols.
A 2007 mechanism:
[Edited on 4-9-2009 by Magpie]
[Edited on 4-9-2009 by Magpie]
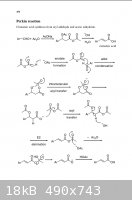
from: http://www.springerlink.com/content/h45v0h1782637462/
[Edited on 4-9-2009 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
hmm jon is interested in cinnamic acid esters also cinnamic anhydride is easily prepared from this by forming the mixed anhydride with acetic
anhydride.
the boiling point of acetic anhydride is very close to the m.p. of cinnamic acid the reaction is driven by distillation of thelower boiling acetic
acid b.p. 118 this may work since the mixed anhydride is a different animal.
the procedure calls for piping the anhydride into heated cinnamic acid.
jon thinks it could be added dropwise or just added in portions.
but propionic anhydride seems prefereable.
since it's b.p is 172 and propionic acid boils at 130~ or so the points are broader and the acid distills closer to the m.p. of cinnamic acid.
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
Magpie, your yield is much better than mine was. I followed the procedure in Vogel 4th edition on a scale of 21 gm benzaldehyde. I used NaOAc
(purchased, anhydrous) instead of KOAc. All reagents were purchased commercially. The procedure looks very much like yours, but there is no
pyridine. I can't tell if you used stirring during the reflux, I suppose you did; I did stir it magnetically. My notes say the reaction turned
yellow, not dark brown, for what its worth. My notes don't mention how much tar was produced, but it was filtered, per Vogel, to remove "resinous
byproducts".
After recrys. from H2O/EtOH, the mp was low (124 vs 133) so the recrys. was repeated using only water. Yield was 2.8 g (9.6%) and mp = 131-132 C.
Vogel gives the expected yield as 18 g (62%).
All in all, I think you did a great job. This prep is a Dog.
|
|
|
crazyboy
Hazard to Others
Posts: 436
Registered: 31-1-2008
Member Is Offline
Mood: Marginally insane
|
|
Anything interesting that cinnamic acid can be used for? Google didn't yield much.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
re: entropy51
Thanks for the comparison of results. As you can see my reaction mix wasn't just yellow, it was dark brown. What amazed me is how fast the tar
formed even at 145C. But it probably doesn't take much to cause heavy coloration.
No, I had no stirring of the reactants in the test tube. A stir bar was used to keep the silicone oil temperature uniform. Stirring of the reactants
looks like a good idea although Brewster did not call for it.
I agree that this procedure has a canine pedigree.  It was probably my dirtiest
synthesis ever. But it did give some product. It was probably my dirtiest
synthesis ever. But it did give some product.
Did your NaOAc give the appearance of not dissolving?
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
Brewster didn't mention stirring because college labs didn't have magnetic stirrers in those days. But you're too young to remember that.
My notes didn't mention anything about the NaOAc not dissolving, but it's been too long for me to remember. I probably would have noted it if that
had happened.
From my perspective, you did a fine job. With stirring you might get even more.
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
that's a tough mechanism to pick apart.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
| Quote: |
Brewster didn't mention stirring because college labs didn't have magnetic stirrers in those days. But you're too young to remember that.
|
That's true and was my guess too. I wonder what Perkin used. We didn't have ground glass fittings either - just bored stoppers.
I checked my Vogel and it specifies 12g of NaOAc. This is the same amount that Brewster specifies yet the benzaldehyde and Ac2O are about half those
of Vogel. That may be an error in scaling down and would explain what appeared to be a gross quantity of NaOAc.
jon: Right, I gave up on trying to understand it after about the first line or so. But I thought I should present what appears to be the latest
thinking on the Perkin reaction.
[Edited on 5-9-2009 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
I'll bet you didn't have heating mantles either. We used bunsen burners for everything in my organic class. Surprisingly we never had an ether fire,
but we did have one benzen fire during the two semester course. The TA put the fire out in about 30 seconds.
When we did Grignards we flame dried the glassware. Before they brought the ether in the TA went around and confiscated all the burners. Worked
quite well to prevent accidents.
|
|
|
Everado E. Dinavo
Harmless
Posts: 15
Registered: 13-8-2009
Member Is Offline
Mood: No Mood
|
|
Cinnamaldehyde is also oxidized by atmospheric O2 to cinnamic acid rather rapidly. I have observed this occur on glasswhere where some cinnamaldehyde
traces were left on overnight, resulting in pale yellow, nearly white crystals with a much different odor. Also, the lid of cinnamaldehyde bottles can
be found to have these crystals and the odor if they have been aged for a few years. I am fairly certain this is not the aldehyde hydrate that is
forming, because I have specifically read that cinnamaldehyde needs to be stored away from oxygen to prevent oxidation. I never actually tried to
identify the crystals, but I might give it another try since it requires about no work, provided you already have the cinnamaldehyde (which you say
you have, Lilly). Just pour out a thin layer of cinnemaldehyde into the bottom of a beaker or petri dish, or even a plate and just leave it until it
turns into crystals. The conversion should be good as long as the surface area is high.
[Edited on 9/10/2009 by Everado E. Dinavo]
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Got aniline? Under the proper conditions, Diazonium Salts, couple nicely with acrylic acid esters, to form cinnamic acid esters. Good yields.
True, aniline is impressively toxic, but reactions of this type, can be used to produce materials, that are very interesting, and very hard to produce
by other methods.
|
|
|
Sleipnir
Harmless
Posts: 8
Registered: 10-9-2009
Member Is Offline
Mood: No Mood
|
|
maybe a oxidation with hydrogen peroxide could be appropriate like in "Hydrogen peroxide oxidation of aldehydes to carboxylic acids: an organic
solvent-, halide- and metal-free procedure "
by Kazuhiko Sato, Mamoru Hyodo, Junko Takagi, Masao Aoki and Ryoji Noyori
(can't read the paper actually)
since i haven't got malonic acid i like the idea of making cinnamic acid from cinnamaldehyde ...
[Edited on 11-9-2009 by Sleipnir]
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
On the other hand, utilizing the diazotization of the amino-acid Phenylalanine, you could form either the halo-acid, or the hydroxy-acid.
Upon dehydration, or de-hydro-halogenation, cinnamic acid should be produced.
This might appear to be an expensive method for producing cinnamic acid.
But, if feasible, there is a rationale for the approach.
In this era, of difficult reagent accessibility, Phenylalanine is very easy to acquire.
There are no shipping problems. And, it isn't very expensive. With almost no effort, I was able to find DL Phenylalanine for sale, for
39.99/Kilo or 15.00/300grams.
[Edited on 12-9-2009 by zed]
|
|
|
JohnWW
International Hazard
Posts: 2849
Registered: 27-7-2004
Location: New Zealand
Member Is Offline
Mood: No Mood
|
|
By DL-phenylalanine, I presume you mean the racemic mixture of its two stereoisomers, marketed as a nutritional supplement for its supposed analgesic
and antidepressant activities, which must be produced synthetically as only the L-stereoisomer occurs naturally as one of the about 20 essential
amino-acids constituting natural proteins. I am somewhat surprised that it is still so readily available, though, because of its potential for
conversion to phenethylamine, and possibilities for conversion of this by methyl or methoxy etc. substitutions to amphetamine or methamphetamine or
related psychoactive compounds.
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Phenylalanine is not easily convertible to any amphetamine. Though it can be done.
It is however, readily convertible to phenethylamine.
Fortunately, most chemists have chosen to NOT pursue, the unenlightened path, of phenethylamine based analgesic synthesis.
More specific to this thread, Phenylalanine might serve as a practical starting material for a fairly simple cinnamic acid synthesis.
Phenylalanine is readily available, at least, here in the States, and the acquisition of reagents like Benzaldehyde or Acetic Anhydride, can be fairly
difficult .
[Edited on 12-9-2009 by zed]
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I recently conducted two experiments with the Perkin Reaction. I attempted to synthesize cinnamic acid from benzaldehyde, acetic anhydride and
triethylamine as a base. 10ml of benzaldehyse, 15ml acetic anhydride and 7 ml of triethylamine where heated together with stirring in a 100ml rbf with
condenser for 3 hours. The mixture was then added to water, steam distilled and a large amount of unreacted benzaldehyde was recovered and used for a
synthesis of alpha phenyl cinnamic acid. The resulting residue in the still pot was boiled with decolorizing charcoal, filtered and strongly acidified
with 12m HCl. After cooling, 0.24g of producted crystallized, for a incredible percentage yield of 2%! I have not taken the Mp yet.
My synthesis of alpha cinnamic acid was much for successful.
5.0g of phenyl acetic acid, awful smell, 6.0 ml benzaldehyde, 4.0ml of triethylamine and 4.0ml of acetic anhydride were heated together in a 50ml rbf
with condenser for 1.5 hours. The work up was according to the procedure specified in Williamson, Microscale and Macroscale Organic Experiments, since
it is to long winded to repeat at this moment. 3.56g of product was obtained in a percent yield of 47%, I have both E and Z isomers isolated according
to the procedure, I still need to take melting points.
[Edited on 17-9-2009 by benzylchloride1]
Amateur NMR spectroscopist
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
My experience:
Benzaldehyde 63 gr,90 gr Ac2O, 33gr Sodium acetate ,2gr pyridine
reaction time:6hour
Yield: 58gr impure(30gr pure)
According to Vogel 5th.experiment 6.138.page1038








[Edited on 24-1-2014 by Waffles SS]
|
|
|
DraconicAcid
International Hazard
Posts: 4332
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Cinnamic acid is nicely oxidized by chlorite; you just have to have a scavenger in there to make sure any hypochlorite that is generated doesn't
attack the double bond or chlorinate the ring.
I took 6.3 mL of cinnimaldehyde, 1.6 g sodium dihydrogen phosphate, dissolved them in 20 mL water and 30 mL isopropanol, and added 6 mL of 30%
hydrogen peroxide. In a separate flask, I dissolved 8.0 g sodium chlorite in 50 mL water, and slowly added this, with stirring to the first solution.
I let it stir an hour (it warmed up, and gave an orange oil that floated on top). After cooling well in an ice bath, I got 6.58 g of off-white
crystals (89% yield), mp 131.5-133.2 oC).
Recrystallizing it from isopropanol (3.8 g in 20 mL, heat to dissolve, cool in ice to precipitate) gave a much smaller yield (0.9 g) of white crystals
(mp 132.2 - 133.9 oC).
I tried a similar trial with a few drops of phosphoric acid added, thinking that a slightly lower pH would speed up the reaction. It gave similar
yield and mp, but the initial foaming and evolution of an orange-yellow gas put me off of trying that again.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
If you're following a literature procedure please provide a reference. Do you really think you need so much hydrogen peroxide as a scavenger in the
Pinnick oxidation?
Chlorite reacts with acid to liberate ClO2, hence the yellow-orange gas you observed with phosphoric acid.
|
|
|
DraconicAcid
International Hazard
Posts: 4332
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
Quote: Originally posted by DJF90  | | If you're following a literature procedure please provide a reference. Do you really think you need so much hydrogen peroxide as a scavenger in the
Pinnick oxidation? |
I'm not an organic chemist- I was following this patent:
http://www.freepatentsonline.com/4549025.html
If you've got a better reference, I'd be delighted to read it.
| Quote: | | Chlorite reacts with acid to liberate ClO2, hence the yellow-orange gas you observed with phosphoric acid. |
Yes, I figured that out myself, thanks.
[Edited on 25-1-2014 by DraconicAcid]
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Hoffnung
Harmless
Posts: 5
Registered: 15-8-2013
Member Is Offline
Mood: No Mood
|
|
Hi all,
I will be also interested in preparing cinnamic acid from cinnamaldehyde soon, surely the next Spring. Since I don't have chlorites I am looking up
for feasible alternatives. I wonder if anyone have tried this one yet:
http://www.freepatentsonline.com/4778924.html
The 8 h of reaction time is discouraging, but I guess that 40ºC is more or less the same as leaving it on the sun in Summer, so it looks interesting.
I'm looking for a reference on the method that Everado E. Dinavo said but I can't find it.
Another method I found in a book is the oxidation using the Tollens reagent, but there aren't any details/recipe.
Well, I'm posting my method and results as soon as I make it. See you.
|
|
|
| Pages:
1
2 |








