| Pages:
1
2 |
Sauron
International Hazard

Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
I would conclude then that it is not s-trithiane.
Perhaps some linear polymer [-CH2-S-]n
Have a look at "Formaldehyde" ACS monograph in forum library pp 129-130
These might be partially sulfurated methylene glycols, in which case treatment with conc HCl will convert them to trithiane
Also, has anyone looked at L.Vanino, Ber 35, 3251 (1902)? Ref 109 from that chapter of the ACS monograph and another thiosulfate prep from formalin
and conc CHl
Worth a look.
[Edited on 19-12-2008 by Sauron]
[Edited on 19-12-2008 by Sauron]
Attachment: Pages from formaldehyde.pdf (822kB)
This file has been downloaded 1066 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
I obtained the original 1902 Ber. paper by same author (L.Vanino) as the 1908 J.Prakt Chem. article translated above.
Main difference is that in his original prep Vanino used a larger volume of 3.5% HCl rather than a smaller volume of conc HCl.
Given the resulting unstirrable glop that results from the 1908 procedure this makes sense.
May be well worth a try.
200 g sodium thiosulfate iun 1 L water
200 g 40% formalin
2 L 3.5% hydrochloric acid
Paper (in German) is attached.
[Edited on 21-12-2008 by Sauron]
Attachment: vanino.pdf (137kB)
This file has been downloaded 721 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
To quote from the ACS monograph Sauron attached above:
| Quote: | Crystalline hydrogen sulfide-formaldehyde reaction products known as
formthionals are also obtained by saturating 37 per cent formaldehyde with
hydrogen sulfide at temperatures in the neighborhood of 40 to 50°C.
Heat is evolved by the reaction and product is precipitated, almost completely
solidifying the reaction mixture. A product of this type obtained
from neutral formaldehyde melted at 80°C after crystallization from chloroform
and contained 51.5 per cent sulfur. When pure, these compounds
are colorless and have little odor.
Under strongly acidic conditions, trithiane or trithioformaldehyde,Sulfuretted FORMALDEHYDE
methylene glycol derivatives of the type described above are converted to
trithiane on heating with concentrated hydrochloric acid. Trithiane is
also produced by reaction of 37 per cent formaldehyde, sodium thiosulfate,
and hydrochloric acid. |
I guess this is the answer then, you just need to use way more acid to force the reaction toward trithiane...which should be soluble in acetone btw.
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
You could add a little activated carbon to the formaldehyde solution in an attempt to remove the blue dye.
| Quote: |
Under strongly acidic conditions, trithiane or trithioformaldehyde,
(CH2S)3, is the principal reaction product of formaldehyde and hydrogen
sulfide and is precipitated in practically quantitative yield.
Trithiane is
also produced by reaction of 37 per cent formaldehyde, sodium thiosulfate,
and hydrochloric acid
|
It does not state that the second reaction needs strongly acidic conditions. Therefore using a larger quantity of lower concentrated acid may work as
Sauron has suggested, and is likely to be preferable as there is more solvent to suspend the product in, reducing tendancy to be left with a
solidified reaction mixture (again as Sauron has suggested).
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
I guess one could infer that the reaction with thiosulfate rather than H2S also needs strongly acidic conditions, which is what I did, but you are
right, it doesn't explicitly say so.
I wonder how the reaction proceeds with thiosulfate: normally it is decomposed to SO2 and S (precip) in the presence of strong acid. One should be
able to smell the SO2 if this reaction was truly taking place. Mechanistically it's difficult to see how elemental sulfur reactions with CH2O...
Alternatively the reaction proceeds by the free H2S2O3 reacting straight with the carbonyl (which is probably activated by the acid) before it can
decompose. Thus sulfate c/would be the product. Therefore the added acid remains, while in the former case it is neutralised as most SO2 will
evaporate.
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
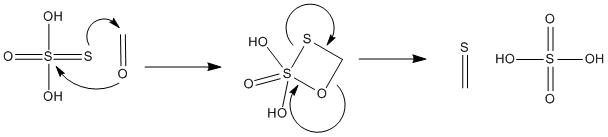
IMO there is no question thiosulfuric acid is the nucleophile which attacks formaldehyde/methyleneglycol. Thiosulfuric acid is a pretty strong
nucleophile...I have attached a proposed mechanism below (I used formaldehyde for simplicity a similar mech could also be developed for methylene
glycol). Also thiosulfuric acid is somewhat long lived (especially in the cold), it takes 15-30 sec after mixing hypo and acid to precip. the sulfur.
Which reminds me, one possible mistake I made was performing this reaction on a still slightly warm formaldehyde solution.
I will not attempt to purify the paraformaldehyde as I have already ordered formalin.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Chemoleo, see Vanino's 1902 paper in which he used dilute rather than concentrated HCL with formalin and thiosulfate and obtained same product in same
(near quantitative) yield.
I still have not seen a complete equation for this reaction and therefore can't draw any conclusions about stoichiometry, mass balance etc.
3 CH2O + 3 Na2S2O3 + 6 HCl -> (CH2S)3 + 3 H2SO4 + 6 NaCl
which looks like the overall reaction.
It is not possible to reconcile the procedure in the 1908 Vanino paper because it is imprecise.
He states "concentrated solution of sodium thiosulfate" 250 g"
"150 cc formalin (concentration not given)
150 cc conc HCL" (not further specified either % or density)
The 1902 Vanino paper from Ber. 35 pp 3251-3252) is on this score, firmer ground
200 g sodium thiosulfate in 1 L water
200 g 40% formalin
2 L 3.5% HCl
Thiosulfate is 158.09 g/mol anhydrous so 1.25 mol or, 0.8 mol if he used the pentahydrate.
Formaldehyde is MW 30 so 80 G is 2.67 mols. That's a largish excess even assuming that the 40% was nominal, and actual wt% was somewhere 35+.
2L of 3.5% HCl is about same as 200 ml 35% which is close to usual conc acid.
So the amount is about the correct one for the purpose of liberating H2S2O3 from sodium thiosulfate. ACS conc HCl is almost 12 M.
[Edited on 21-12-2008 by Sauron]
Sic gorgeamus a los subjectatus nunc.
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Yes, this checks out. Smuv's only difference is that he used 100 ml of conc Hcl rather than 1 litre of 1:10 HCl.
Another question - is the conversion of paraformaldehyde to formaldehyde quantitative under these conditions? There are no lower polymers that are
soluble?
Smuv, very nice the mechanism, you beat me to it, this is more or less what I had in mind, including the formation of SO4(2-).
I wonder if the reaction would be possible by using i.e. Al2S3 (from Al and S powders). Since Al2S3 is water-labile, it would have to be added in
small portions to the CH2O/HCl solution. Better yet, use FeS, and use acid addition to liberate the H2S.
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
My Way: Thiocyanic Acid
Preface: Based upon the mechanism I developed above, I theorized that other reagents with nucleophilic sulfur synthons might be able
to react analogously. The two candidates were thiourea and thiocyanate. I gave thiocyanate a shot because I had it on hand and I thought
polymerization might be a side reaction with thiourea. Of course, the advantage of this is that there is no chance of sulfur being precipitated.
Experimental
Formaldehyde solution
1g Paraformaldehyde (most of blue dye was removed by washing with alcohol...forget which one)
2.5mL Water
NaOH (catalytic amt)
Upon heating + swirling ca. .1g did not dissolve, so the solution was vac filtered to remove insolubles.
1,3,5-Trithiane
Formaldehyde Sol from above
1.5g Ammonium Thiocyanate
3.0mL HCl
The ammonium thiocyanate (once anhydrous ) was dissolved into the formaldehyde sol.
(at ca. 10c). The HCl was then added, the solution slowly precipitated very fine crystals over the course of a few minutes. The solution was vac
filtered, yielding beautiful tiny light blue needles (filtrate boiled in filter flask durring vac filtration...isocyanic acid?). The filter cake did
not readily dry by passing air through it at the pump, so it was washed with ~2mL of isopropanol (if only I had it on hand I would have used ether).
The isopropanol unfortunately dissolved ca 50% of the product leaving a nearly white non-crystalline (odd!) powder. ) was dissolved into the formaldehyde sol.
(at ca. 10c). The HCl was then added, the solution slowly precipitated very fine crystals over the course of a few minutes. The solution was vac
filtered, yielding beautiful tiny light blue needles (filtrate boiled in filter flask durring vac filtration...isocyanic acid?). The filter cake did
not readily dry by passing air through it at the pump, so it was washed with ~2mL of isopropanol (if only I had it on hand I would have used ether).
The isopropanol unfortunately dissolved ca 50% of the product leaving a nearly white non-crystalline (odd!) powder.

(Filter cake before washing with isopropanol, red spots are probably due to reaction between my damn ferrous spatula and thiocyanate.)
The melting point of the powder was wide around 212C (sorry I had trouble viewing the thing while it melted, but it was definitely in-between states
at 212) with decomp to a brown red liquid. One thing to note...the crystals were not fully dry and melted/dehydrated around 80c. I am sorry my
melting point results are so shitty, I am having a lot of trouble with the equipment I have available (and my frigid fingers).
Discussion
The product obtained from this process smells just like the product from the thiosulfate procedure. The smell is hard to describe, something like the
smell of sulfur mixed with formaldehyde.
The reason the product was not dried in an oven was because I tried this with the recrystalized product from the thiosulfate procedure and half of the
crystals sublimed after 15min at 150F in a convection oven.
I am not going to be quick to label this product as trithiane (although I believe it is) before repeating this procedure on a commercial formalin
solution and obtaining the mp on a dry product.
No matter what...I am happy
@Chemoleo, methylene glycol(s) are the predominant species in an aqueous formaldehyde solution. This begs the question...why did I start from
formaldehyde in my mechanism above? I was lazy and wanted to convey the gist of the mechanism with the fewest number of arrows possible.
[Edited on 12-21-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Somewhere in the lit., very likely in that ACS monograph on HCHO cemistry, there will be something on the reaction of thiocyanates or thiocyanic acid
and formalin.
As noted upthread, KSCN reacts with Zn and HCl to give trithiane. Ammonium thiocyanate is a lot costlier than potassium thiocyanate.
As to the paraformaldehyde conversion, assuming that the depolymerization goes to completion and at that point there is just hydrated formaldehyde
(methylene glycol) and water, sans methanol stabilizer, the process of repolymerization will commence and the soln will cloud. I suppose that can be
followed instrumentally. For purposes of the trithiane prep I doubt that it matters. Vanino always has formalin in about 2X excess anyway.
Sic gorgeamus a los subjectatus nunc.
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
| Quote: | | As noted upthread, KSCN reacts with Zn and HCl to give trithiane. |
This is a completely different method. While I don't currently have access to the paper, looking at the reagents it is clear to me that Hofmann was
trying to generate hydrogen sulfide in situ by reducing thiocyanate with zinc.
The relavent equations would be:
1. Zn + SCN- --> CN- + S-2
2. S-2 + 2H+ --> H2S
3. 3HCHO + 3H2S --> 1Trithiane + 3H2O
vs my method:
1. 3SCN- + 3HCHO --> 1Trithiane + 3OCN-
| Quote: | | Ammonium thiocyanate is a lot costlier than potassium thiocyanate. |
If you do it you may use any salt you like. I think though even on a molar scale ammonium thiocyanate wont break the bank.
| Quote: |
Somewhere in the lit., very likely in that ACS monograph on HCHO cemistry, there will be something on the reaction of thiocyanates or thiocyanic acid
and formalin. |
In the ACS article there is something about polymer formation in neutral media upon reflux.
If you want to discus these points further, lets do it over PM, as I don't want to further side-track the thread.
[Edited on 12-21-2008 by smuv]
[Edited on 12-22-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Oh no, this is interesting to everyone, and has bearing on the subject at hand!
I think I'll split the thread into trithiane synthesis and chloromethylsulfonyl chloride later, to keep this aspect tidy - I'll put in a
cross-reference.
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
I obtained the 1868 Ber, paper by Hofmann for comparison of yield. Will report back after ploughing through.
As the thiosulfate is more ubiquitous not to mention cheap, and does not involve H2S generation even in situ, (merely NaCl and maybe Na2SO4) it is far
more attractive. I was just looking to see what sorts of yields these Zn reductions gave. I'd be surprised if they are as efficient as the thiosulfate
(or the H2S) methods which are essentially quantitative.
I am not particularly interested in the halomethylsulfonyl halides at least at present. But s-trithiane is another matter because it, along with
1,3-dithiane, are useful reagents for maskted thioketals useful in general routes to aldehydes. I was poised to make trithiane with H2S but you have
saved me from that. Thanks, and I will certainly try to help in any way I can.
Also saved me from paying $2 a gram for this stuff....
[Edited on 22-12-2008 by Sauron]
Attachment: HofmannBer1868.pdf (1.4MB)
This file has been downloaded 679 times
Sic gorgeamus a los subjectatus nunc.
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Oddities from all angles
I have done a bit of work on the attempted synthesis of various thio acetals/ketals and their polymers. Although I am confused by the results.
Attempted synthesis of thioacetone/its polymers
2.0mL acetone (27.2mmol)
1.5g Ammonium Thiocyanate (19.7mmol)
4.0mL 31.45% HCl (excess)
A mixture of ammonium thiocyanate and acetone was swirled (not much ammonium thiocyanate dissolved). Next the HCl was added, immediately depositing a
white non-crystalline precipitate. The solution was allowed to stand for a few minutes and was vac filtered with the filtrate used to complete the
transfer. The filter cake was dried at the pump; Yield .8g.

(The red is again to due my ferrous spatula. The stain had almost completely faded after standing overnight.)
The product sublimed before melting, very rapidly around 295c. Heating the product in a capillary tube with a free flame melted the product to a
clear white liquid with vigorous decomposition. A sample of the product heated by naked flame on the tip of a spatula does not sustain its own
combustion, but slowly sublimes/decomposes (without melting) with a sulfury smell, leaving no residue. The product was appreciably water soluble.
have no idea what it is...
Attempted Thiobenzaldehyde/polymers
10mL of McCormick Immatation almond Extract
6mL 31.45% HCl
1.5g Ammonium Thiocyanate
The ammonium thiocyanate was mostly dissolved in the almond extract and then HCl was added; an oily layer (benzaldehyde) formed. The mixture was
magnetically stirred for 30 min, yielding no solid precip.
Attempted Trithiane From Thiosulfate
7g Sodium Thiosulfaate
7mL 37% Formalin
7mL 31.45% HCl
Most of the thiosulfate was dissolved into the formalin; Next the HCl was added with magnetic stirring. After ca. 1 minute magnetic stirring was
stopped and ~30sec later a white precipitate spontanously came out of solution with the evolution of a good deal of heat. The solution was allowed to
stand for a few minutes and vac filtered with the filtrate used to complete the transfer. The filter cake was washed with 2x 5mL of water; almost all
of the product dissolved! The filtrate was perfectly clear and non-turbid. WTF!
Attempted Trithiane from Thiocyanate
3.6g Ammonium Thiocyanate
7mL 37% of Formalin
7mL of 31.4% HCl
Most of the ammonium thiocyanate was dissolved into the formalin with stirring. Next, the HCl was added, immediately forming a white, non-crystalline
precipitate. The solution was allowed to stand for ~40min and vac filtered, with the filtrate used to complete the transfer. The filter cake was
washed w/ 2x3mL water; almost all of it dissolved! The filtrate was slightly turbid, but no where near turbid enough to account for the loss of
almost the entire product.
Discussion
These results have left me scratching my head, I don't understand why my products all come out being very water soluble, this makes absolutely no
sense to me (I need to drink on it).
You can view the data about the formalin I bought. The acetone and hydrochloric acid were bought at a hardware store. The thiosulfate and thiocyanate were photo
grade.
The benzaldehyde may not have reacted like acetone and formaldehyde because it is a much weaker electrophile.
[Edited on 12-22-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Very Interesting!
Excuse the double post but this is such an interesting development I feel it is worthy of its own post.
The filtrate from the attempted preparation of sym-trithiane was allowed to stand ca. 20h (if you recall the filter cake dissolved in water upon
washing). Upon standing a heavy yellow oil precipitated. When I first encountered it my first reaction was to smell it (really terrible lab
etiquette), it had an amazingly piercing smell of mustard. I believe this oil to be a methylene isothiocyanate derivative (could be somewhat
polymeric, SCN-C-(O-CH2-O)n-CH2-NCS, the thio derivative or could be HO-CH2-NCS) or possibly a methylene
thiocyanate derivative.

(picture of oil with flash, the oil is easy to see but the colors are inaccurate)

(picture of oil w/o flash, colors are accurate but the oil is hard to see)
I am debating whether to further work-up this product as since I don't know what it is it is hard to gauge the toxicity. I would assume though that
isothiocyanates shouldn't be TOO toxic, although highly irritating to work with.
--
Late EDIT: Finally and Definitively Success!
So, I have tracked down the source of my strange results: my very cold lab. My lab is in a cold basement, the temperature hovers between 8 and 10c.
I believe the products from my experiments 1 post ago were skewed because of the cool temperatures. In order to investigate the effect of temperature
upon this reaction, I tried the synthesis of trithiane from formalin and thiosulfate at 85c.
Synthesis of sym-trithiane
7g Sodium Thiosulfate (anhy.)
7mL 31.45% Hydrochloric Acid
7mL 37% Formalin
The sodium thiosulfate was dissolved into the formalin at 85c (reflux). Next, the solution was removed from the hotplate and the HCl was added.
After addition, the solution immediately began to vigorously boil/evolve a gas and formed a white/yellow precipitate. There was a distinct, heavy,
sulfury smell, but no smell of SO2 was noted. The solution was allowed to cool to ca. 20c and vac filtered with the filtrate used to complete the
transfer. The filter cake was washed w/ 2x ~4mL water, dissolving a good amount of inorganic salts. After washing a chunky slightly sulfur tinted
product was obtained. The contents of the filter cake were crushed with a mortar and pestle and again washed with 4mL of water. A fluffy, light
yellow, slightly damp powder was obtained; yield 2.0g.
About half of the crude product was subjected to further purification. Therefore, ca. 1g of product was dissolved into a minimal quantity of boiling
AcOH. Insolubles were removed by vac filtration, causing much product to spontaneously precipitate in the filter funnel and filter flask. The
filtrate/precip in the filter flask was heated to boiling and slowly cooled over the course of ~30min; a very fine sand of white crystals was
deposited. The solution was quantitatively vac filtered and the crystals were dried somewhat in the filter funnel. The crystals were then
recrystallized from boiling toluene, allowed to cool to room temp, then allowed to cool outside in a snow bath to ca. -6C. The crystals were
recovered via vac filtration and dried in the filter funnel. White, ~4mm long crystalline needles were obtained; mp 209-210. The crystals melted w/o
decomp. into a clear liquid.

(On Left: About Half of the crude product; On Right: Recrystallized product.)

(Pretty little crystals)
Its done, I have trithiane!
Some Things
It looks like the yield of at least crude trithiane is nearly quantitative. The crude product definitely has a little sulfur mixed in, although upon
melting forms a clear liquid w/ no red color from sulfur. I don't think reflux is the best temp to do this reaction (because of more sulfur
formation), but it was an effective way of determining the effect of temperature upon the reaction.
I would like to point out, that my purification was oriented towards quality not quantity. Therefore, I accepted the fact that during the AcOH
extraction, most of the product recrystallized on the filter paper (despite the funnel being pre-heated with a heat gun).
I believe the recovered precipitates from my experiments 1 post ago were largely inorganic. I also believe that the filtrates contained free
thiosulfuric/(iso)thiocyanic acid and HO-CH2-X (where X is SCN or S=SO3H). In the case of the oil obtained, HO-CH2-SCN probably disproportionated to
NCS-CH2-SCN and formaldehyde. I do not believe the oil is SCN-CH2-NCS, because I would expect this compound to be a solid.
[Edited on 12-24-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Beautifull! Well done!
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Bravo!
Cold lab I don't need to worry about in Thailand.
Sic gorgeamus a los subjectatus nunc.
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Investigation: Cold Temps
I wanted to know what was going on at low temperatures, in the reaction between formaldehyde and thiosulfuric acid, so of course I did a few
experiments. 
I tried the synthesis of trithiane at ice bath temperatures (using the same ratio of reagents as i have been using, 1:1:1). After mixing the reagents
a precipitate formed (inorganic crap). The solution was allowed to stand for a few min, and was vac filtered. The filtrate was allowed to stand for
ca. 3.5 hours at 12c and the white precipitate which formed was vac filtered. The filtrate was again allowed to stand. Over the course of another
day 2 more crops of precipitate were collected. The total mass of precip was way greater than the theoretical yield of trithiane. The percipitate was
a fine, pure-white, semi-crystalline powder, with a mp >260c and burned without residue. I believe this is a thiomethylene-oxymethylene polymer.
I also theorized that at cool temps S-hydroxymethylthiosulfuric acid was the intermediate metastable product. If this was the case, I hoped that this
intermediate might be able to be chlorinated to form chloromethanesulfonyl chloride (like a bunte salt, followed by the nucleophilic attack of the
intermediate hydroxymethanesulfonylchloride by chloride). Therefore, at ice bath temps (with pre-cooled reagents), stoichiometric quantities of
formalin, hydrochloric acid and sodium thiosulfate were mixed. The precip (inorganic) was removed via vac filtration and the filtrate was chlorinated
with ice cooling by adding 3eq tcca portionwise. A good deal of heat was evolved with each addition. After addition the solution was stirred for an
hour out of the ice bath. The cyanuric acid/sulfur precip was removed via vac filtration yeilding a bright yellow-green filtrate. A large excess of
conc. HCl was added to the filtrate, with the evolution of little/no elemental chlorine. After standing a few hours nothing really happened. Based
upon the production of elemental sulfur during the chlorination, I theorize (albeit based on slim evidence) that the S-hydroxymethylthiosulfuric acid
adduct is not a long lived intermediate with respect to disproportionation to thiosulfuric acid and formaldehyde. The rise in heat upon addition is
likely due to the reduction of chlorine by thiosulfuric acid.
Just thought I would share; hopefully this stuff is as interesting to some people as it is to me.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Sauron
International Hazard
Posts: 5351
Registered: 22-12-2006
Location: Barad-Dur, Mordor
Member Is Offline
Mood: metastable
|
|
Vanino did say to reflux the mix. In his original prep with lots of water, that would be possible, but there's little chance that the typical
low-silution thick sludge is going to work that way.
Sic gorgeamus a los subjectatus nunc.
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Success!? (more or less)
I finally had success with both synthesis. I worked out the kinks in the trithiane synthesis, to yield a fairly pure crude product. For the
synthesis of chloromethylsulfonyl chloride, despite my best attempts, I only had successes following procedures involving elemental Cl2.
Trithiane Synthesis
30g Sodium thiosulfate
40mL H2O
25mL 37% Formalin
25mL 31.45% HCl
The sodium thiosulfate and water were mixed with stirring; most of the sodium thiosulfate dissolves. Next, the formalin was added, nearly all of the
sodium thiosulfate dissolved. The HCl was subsequently added, causing the solution to immediately turn a turbid white. Under strong magnetic
stirring, the solution was brought as quickly as the hotplate would allow to reflux. Like a shot, at reflux an exothermic reaction set in which
yielded a clumpy white precipitate. The flask was removed from the hotplate, after the exotherm subsided, and allowed to cool to room temp. The
precipitate was collected by vac filtration and thoroughly washed in the filter funnel. After carefully drying the crystals outside on a hot plate
(the crude, wet product smells very thiol-like, H2S?), 8.1g of a white powder with a subtle fowl odor was collected.

(After addition of HCl, (sorry for the blur, I don't understand my auto-focus))

(Durring the exothermic reaction, at reflux)

(The dry product)
The batch depicted here was not characterized, however a previous smaller batch (.25x this batch) was characterized. The crude product, melted with
decomp over a wide rage ca. 170-200c. After one recrystalization from toluene (which dissolved nearly all of the product) the mp narrowed only
slightly. This once recrystallized product was fed into the chloromethylsulfonyl chloride prep.
Chloromethylsulfonyl Chloride
ca 7ml AcOH
ca .75 mL H2O
.5g Trithiane
3g TCCA
20mL HCl
(see apparatus below to better understand the chlorination procedure) The TCCA was added to a filter flask followed by an equal volume of water; next
a balloon and silicone tube were affixed. The HCl was added via a syringe (using the rubber tube), the silicon tube was clamped shut and the syringe
was removed. A modified pipette was affixed to the end of the rubber tube to serve as the gas bubbler. Finally the clamp was removed and
chlorination commenced.

(The beginning of the Chlorination)
From this apparatus chlorine was led into a waterbath-cooled test tube containing the AcOH, trithiane and water. The balloon did not have enough
elasticity to provide adequete pressure throughout the experiment, so I had to squeeze it to feed the last 50% of chlorine. The pipette was swirled
around during the chlorination (ugh!, wrist got tired). The chlorination lasted ca. 45 min. The AcOH mixture was originally a suspension, but as the
chlorination proceeded, more trithiane was taken into solution. The chlorination was moderately exothermic. After chlorination the solution was
allowed to stand for ~30 min.

(Solution after standing, post chlorination)

(The oil)
After standing, the contents of the test tube were poured onto crushed ice and water was added, precipitating a clear, slightly yellow oil.
I assume this oil to be chloromethylsulfonyl chloride. I will not be able to characterize this product for many months unfortunately.
Failed in situ Halogenations
AcOH, NH4C,l TCCA (non-)Chlorinating system
.5g Trithiane
6mL AcOH
.50mL H2O
1g NH4Cl
3g TCCA
All the reagents, save the TCCA were mixed into a 25mL flask. Under stirring, the TCCA was added, with exotherm upon each addition. The addition
lasted 45min and then the solution was allowed to stir for another 30min. Next, an excess of HCl was added (releasing no Cl2) and the solution was
diluted with ice-water and vac filtered. No oil was noted.
TCCA, NH4Br, AcOH system
.5g Trithiane
8mL AcOH
4g TCCA
.50mL H2O
2.5g NH4Br
In an ice bath, the water, AcOH and ammonium bromide were mixed together; ca. half of the ammonium bromide went into solution. The TCCA was added in
portions with magnetic stirring, this was slightly exothermic. To the dark red-orange solution which resulted, trithiane was added in portions over
45 min. Each addition resulted in an exotherm. The solution was allowed to stir for 45 min and stand for another 30 min. Upon workup no oil came
out of solution.
Discussion
This is all bitter sweet. While I seemed to have success with the direct chlorination I never go the in situ process worked out. Also, I never got
the chance to characterize the product (or see how its esters alkylate)
The chlorination system, of TCCA, NH4Cl and AcOH was pretty well mannered, with very little evolution Cl2. There was definitely some reaction taking
place, because TCCA does not release much/any heat upon addition to acid. I wonder if i should have let the thing go longer?
The ammonium bromide trial, was sort of a last ditch effort, to get a product from the in situ halogenation. I was guessing that bromomethanesulfonyl
bromide would be the major product (but what to I know). I was hoping the production of bromine before addition of the trithiane would help determine
if the cyanuric acid was reacting with the sulfonyl halide at all. However, the way the system worked out, the bromine was not produced all at once,
but slowly throughout the reaction. After each addition the color would lighten somewhat, and after some time stirring darken to some steady color
once again.
About the direct chlorination, maybe my declaration of success is a little premature, but whatever...I need closure.
That's all Folks; wont be able to work on any more of this for a at least a few months.
[Edited on 12-30-2008 by smuv]
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
An oxidation method for chlorinating thiols to alkanesulfonyl chlorides using TCCA in aqueous acetonitrile has been recently reported on the Synthetic
pages:
Oxidation of a thiol to a sulfonyl chloride; Dodecane-1-sulfonyl chloride
This is in support to smuv's idea above and might also be a practical method for those who want to prepare other sulfonyl chlorides from the
relatively easily accessible thiols/thicyanates/isothioureas/thiosulfates/etc.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Fery
International Hazard
Posts: 1026
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
I tried the 1,3,5 trithiane synthesis with these steps:
expected equation:
3 CH2O + 3 Na2S2O3 + 3 HCl -> (CH2S)3 + 3 NaCl + 3 NaHSO4
formaldehyd M=30,03 g/mol 37% density 1,09 g/cm3
Na2S2O3 . 5 H2O M = 248.18 g/mol (pentahydrate) M=158,11 g/mol (anhydrous)
HCl M=36,46 g/mol density 35% HCl = 1,18 g/cm3
0,50 mol scale:
formaldehyd 15,0 g 100% = 39,5 g 38% = 36,25 cm3 (density 1,09 g/cm3)
Na2S2O3 . 5 H2O = 124,1 g or 79,1 g (anhydrous 158.11 g/mol)
HCl = 18,23 g 100% = 51,1 g 35% = 44,1 cm3 (density 1,18 g/cm3)
130,2 g Na2S2O3 . 5 H2O (slight excess circa 5%) dissolved in 150 g H2O in 1 L 3-neck RBF. All thiosulfate dissolved (endothermic, so then warmed to
room temperature). 39,5 g 38% formaldehyde added at room temperature (yes weight on scale, I do not measure volumes). Thermometer in 1 neck, dropping
funnel in second neck, condenser in third neck, the top of condenser connected to a hose to lead all gases outside of lab.
Content in flask warmed to 60 C on oil bath at magnetic stirred, hotplate heating turned off and started addition of 51,1 g 35% HCl from dropping
funnel, T raised to 85 C due to accumulated heat in hotplate and reaction temperature. The temp was kept at 85 C, HCl drop rate 1 drop per second,
addition lasted 20 minutes, then heating turned on, refluxing started in 15 minutes and then refluxed for 30 minutes (T 103 C). Then let to cool down
to room temperature overnight.
Gravity filtered, washed with 25 ml water on filter 2 times.
(filtrate treated the same way with fresh 61,1 g 35% HCl but now no more product obtained, only some turbidity observed in the reaction flask)
Air dried for 5 days on filter paper, on day 4th and 5th the weight already stable 23,6 g of dry powder.
11,5 g of crude product charged into cellulose extraction thimble and inserted into 100 ml Soxhlet extractor, on top of 500 ml RBF with 210 g of conc.
acetic acid. Performed 25 extraction cycles (circa 2,5 L of acetic acid washed the product) - this solvent proven inefficient, something passed into
RBF but most of the product stayed in the thimble.
11,5 g of the crude product extracted now with circa 150 ml of benzene, performed 25 cycles (circa 2,5 L benzene washed the crude product).
The volume in RBF reduced to circa 80 ml by distilling out of half of the benzene and let to slowly cool to room temperature. Yellow needle crystals
decanted from solvent and air dried on filter paper.
6,9 g of yellow product m.p. 118-121 C = sulfur !!!!
3,8 g of colorless product with unknown formula stayed in the thimble (insoluble in benzene, I do not know whether this is something inorganic trapped
in crystals or adsorbed on crude sulfur product or whether some polymer of formaldehyde???).
So I expect this reaction:
1:1:1
Na2S2O3 + HCl + CH2O -> NaCl + S + HOCH2SO3Na (formaldehyde bisulfite adduct)
Using excess HCl (1:2) could react this way leaving formaldehyde unreacted:
Na2S2O3 + 2 HCl -> 2 NaCl + S + SO2 + H2O
Maybe using less Na2S2O3 thus having more acidic environment could lead to different reaction? Smuv used half of the amount of thiosulfate in his
reactions...
So I will have to try to bubble nasty H2S into the solution of HCl + formaldehyde. I wanted to avoid that by using Na2S2O3 which did not work in my
case (but maybe using less thiosulfate could work???). I have Na2S but it is not colorless, it is in a form of yellow flakes so dropping Na2S solution
into HCl+formaldehyde could introduce disulfide or even polysulfides which would decompose to sulfur and contaminate product. That's why I wont plane
to drip Na2S solution into solution of HCl+formaldehyde but I will use Na2S + extra HCl to generate gaseous H2S which prevents introduction of sulfur
into the reaction (possible H2S2 or H2S3 would decompose in the generator leaving only pure H2S).
|
|
|
| Pages:
1
2 |
|









