RogueRose
International Hazard

Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
40gal Muriatic acid advertised for "free"
Just curious if there is anything that could be done with 40 gals of free Muriatic acid. I know it can be used to make other acid often but is there
anything that you can think of that you would do with a quantity such as this?
|
|
|
unionised
International Hazard
Posts: 5126
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Sell it.
|
|
|
myristicinaldehyde
Hazard to Others
Posts: 166
Registered: 23-4-2016
Location: .͐͌ ͛҉̻̫̰̻̖E̮ͮ̐́̚ ̢̗̅̉ͩ͂̒̌.̯̻̺̯̀̎͂̄ͩ̚
Member Is Offline
Mood: сорок пять
|
|
The acid I can get at the hardware store is shit. (Well, if it for free, perhaps you can send 1L samples  ) )
I'd buy it
[Edited on 2016-6-7 by myristicinaldehyde]
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
Quote: Originally posted by myristicinaldehyde  | The acid I can get at the hardware store is shit. (Well, if it for free, perhaps you can send 1L samples )
I'd buy it
[Edited on 2016-6-7 by myristicinaldehyde] |
Well this is "free" so I'm a little cautious and wondering if it is polluted.
|
|
|
unionised
International Hazard
Posts: 5126
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
At about 20% HCl by weight it distils nicely.
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
Here is what I am thinking: I'm curious if it is true 31.45% HCl or some lesser 12, 15 or 15% HCl. Please excuse this remedial question as I know
the jist of some of this, but verifying it all before this dissappears (business giving it away) may be difficult.
So, can I purify it and concentrate it by bubbling in more Cl?
Next access to Ca(NO3)2, KNO3 and possibly NaNO3 (if the CalNit is converted to NaNO3) isn't much of a problem.
Once the HCl is purified and concentrated I can add a nitrate to get nitric acid
Chlorates may be possible if that would be good to make an acid. Perchlorates are a little more difficult to aquire.
H2SO4 would be my ideal acid next to Nitric or Perchloric so what about adding ammonium sulfate? in the end I shoul have ammonium chloride and H2SO4
in a dilute form, correct (if it is 32% HCl, then I would have about 32% H2SO4 and I would be happy with the ammonium chloride that is left behind.
I am looking for a good source of pure CaCl2 and I have a lot of CaCO3, so I should get CaCl2 + H2O + CO2, correct?
So my biggest question. Let's say I clean it with a SUPER fine filter like .05 micron (yeah I know I hear ya'all) would that help remove any yellow
iron from it?
|
|
|
Metacelsus
International Hazard
Posts: 2539
Registered: 26-12-2012
Location: Boston, MA
Member Is Offline
Mood: Double, double, toil and trouble
|
|
Adding a nitrate salt to HCl gives you a complex mixture akin to aqua regia. You can't make pure nitric acid this way.
Likewise, how do you plan to separate the ammonium chloride from the HCl + ammonium sulfate?
Finally, the iron would be dissolved, and thus impossible to filter out. Distillation would work, however.
|
|
|
Dr.Bob
International Hazard
Posts: 2736
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
I used to get chemicals free all of the time from people who needed to get rid of them and did not want to pay someone to dispose of them as waste.
I would buy it, but only if I could store and use that much safely or find others to share in it.
|
|
|
Richard3050
Harmless
Posts: 17
Registered: 20-3-2016
Member Is Offline
Mood: No Mood
|
|
Now all you need to find is a bunch of free zinc and you can revamp your supply of free zinc chloride and hydrogen gas. Not sure why you need that but
whatever works. I am also curious where you would plan to store 40 gallons of acid.
"Science knowledge only adds to the excitement... I don't understand how it subtracts."
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
| Quote: Originally posted by Richard3050 | | Now all you need to find is a bunch of free zinc and you can revamp your supply of free zinc chloride and hydrogen gas. Not sure why you need that but
whatever works. I am also curious where you would plan to store 40 gallons of acid. |
Same place I keep my 400 gallons of H2O2 J/k!
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
That H2O2 was a joke, it is only H2O unfortunately, that extra 2 makes a little difference doesn't it.
I've seen some methods to get pure HCl (closed tupperware container w/ DH2O container inside). This will give 1/2 the concentration and I've heard
the azeotrope is about 20%.
If distillation is used will it off-gas the Cl first allowing it to be bubbled into a solution of DH2O?
Also, if distillation is used and once liquid starts coming over I am guessing I am going to be getting the 20% azeotrope coming over (after the
initial Cl gases have stopped being emmited). Will any contaminates come over with the gases like when essential oils or distilled spirits (liquor)
brings over flavor?
|
|
|
Texium
Administrator
Posts: 4587
Registered: 11-1-2014
Location: Salt Lake City
Member Is Offline
Mood: PhD candidate!
|
|
Well, first of all, it's not "Cl" gas, it's HCl gas. Hydrochloric acid is HCl gas dissolved in water. There really shouldn't be any other volatile
contaminants, so distilling it should leave you with quite pure azeotropic acid. The main contaminant that will likely be there is iron, and none of
it will carry over.
Also, regarding making nitric acid or sulfuric acid using your hydrochloric acid, neither is feasible. As previously stated, mixing hydrochloric acid
with nitric acid or a nitrate salt will produce aqua regia, and once you've made it there's no going back. Adding hydrochloric acid to a sulfate salt
will appear to do absolutely nothing. This is because there is nothing driving the equilibrium to either side. As there's not really a way to remove
ammonium chloride or sulfuric acid from the mixture without removing ammonium sulfate or hydrochloric acid as well, you won't be able to get anything
out if it. The reverse reaction however is very doable, as HCl can be distilled from the mixture as the only volatile product, which will shift the
equilibrium to be favoring HCl.
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
| Quote: Originally posted by zts16 | Well, first of all, it's not "Cl" gas, it's HCl gas. Hydrochloric acid is HCl gas dissolved in water. There really shouldn't be any other volatile
contaminants, so distilling it should leave you with quite pure azeotropic acid. The main contaminant that will likely be there is iron, and none of
it will carry over.
Also, regarding making nitric acid or sulfuric acid using your hydrochloric acid, neither is feasible. As previously stated, mixing hydrochloric acid
with nitric acid or a nitrate salt will produce aqua regia, and once you've made it there's no going back. Adding hydrochloric acid to a sulfate salt
will appear to do absolutely nothing. This is because there is nothing driving the equilibrium to either side. As there's not really a way to remove
ammonium chloride or sulfuric acid from the mixture without removing ammonium sulfate or hydrochloric acid as well, you won't be able to get anything
out if it. The reverse reaction however is very doable, as HCl can be distilled from the mixture as the only volatile product, which will shift the
equilibrium to be favoring HCl. |
Thank you for the explinations. I read so many posts here over the months and get mixed up as to what is possible vs what people "say" is possible.
If the goal is to get pure HCl w/o any impurities (the standard yellow color or even a slight green tint - iron or possibly copper maybe??) I've read
that putting a gallon of HCl in an open container/tub along with another container/tub with a gallon of DH2O will give pure HCl in the DH2O container
- All must be in an air tight container.
What I'm wondering is if doing something like distilling or simple heating and piping vapors into a column of DH2O to bubble up through.
I see that BP increases as HCl content decreases.
What is the best and also easiest (2 options?) to get a high concentration (30%+) of pure HCl from the muriatic.
I have use for lower concentration HCl from say 5-12% and it doesn't need to be pure, so if the HCl could be evap'd and re-absorbed leaving the lower
concentration as the last 5-12%, that would be the best alternative. Can this be done with a heat distill/evap setup with a bubbler into DH2O?
|
|
|
Deathunter88
National Hazard
Posts: 519
Registered: 20-2-2015
Location: Beijing, China
Member Is Offline
Mood: No Mood
|
|
Assuming your muriatic acid is above 20% I think the following is best:
Dilute some of your acid to a concentration below 20%.
Distill the acid and keep the fraction boiling 107-110˚C (the 20% azeotrope)
Put your distilled acid in a graduated cylinder.
Put the raw >20% acid in a flask with a tube leading into the graduated cylinder with pure acid.
Heat the flask with raw acid until its boiling point reaches 108˚C.
Distill the resulting lower concentration raw acid and repeat the process.
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
| Quote: Originally posted by Deathunter88 | Assuming your muriatic acid is above 20% I think the following is best:
Dilute some of your acid to a concentration below 20%.
Distill the acid and keep the fraction boiling 107-110˚C (the 20% azeotrope)
Put your distilled acid in a graduated cylinder.
Put the raw >20% acid in a flask with a tube leading into the graduated cylinder with pure acid.
Heat the flask with raw acid until its boiling point reaches 108˚C.
Distill the resulting lower concentration raw acid and repeat the process. |
Thanks for the suggestions! That is kind of what I was thinking after reading many posts related to HCl.
I did come across one post that said that by adding CaCl2 (some said MgCl2 is even better) to HCl that this will release a lot of the HCl gas and can
be taken down past the 20% azeotrope. I think this process is exothermic so an icebath is necessary.
I'm still trying to figure out how to deal with such-back and I see the funnel method is supposed to work pretty well.
|
|
|
RogueRose
International Hazard
Posts: 1594
Registered: 16-6-2014
Member Is Offline
|
|
| Quote: Originally posted by Deathunter88 | Assuming your muriatic acid is above 20% I think the following is best:
Dilute some of your acid to a concentration below 20%.
Distill the acid and keep the fraction boiling 107-110˚C (the 20% azeotrope)
Put your distilled acid in a graduated cylinder.
Put the raw >20% acid in a flask with a tube leading into the graduated cylinder with pure acid. Heat the flask with raw acid until its
boiling point reaches 108˚C.
Distill the resulting lower concentration raw acid and repeat the process. |
When the tube is in the cylinder should it be submerged and have some kind of bubbler in it or what is the best way to do this?
|
|
|
Arg0nAddict
Hazard to Self
Posts: 54
Registered: 8-6-2016
Location: PNW
Member Is Offline
Mood: Mercuric
|
|
| Quote: |
I'm still trying to figure out how to deal with such-back and I see the funnel method is supposed to work pretty well. |
Easy problem to solve, if its a sealed glass setup use a suckback stop, its like a one way valve.
If you don't want to buy anything you can use empty flask in the center. This is how I bubble a few gases anyway; H2S, chlorine and acetylene...
Don't just use a open tube because you will waste way more gas with such little surface area. You want tons of small bubbles going slowly. I have
found that a fish tank bubble maker going into a graduated cylinder works best because the bubbles can rise and be exposed more.
A bit of warning I am all self taught, so im sorry if I am not using the right terminology here.
Pro Tip: Don't seal the last piece of glassware, I had a cyanide volcano happen on accident. Well I loosely set a stopper there so the glass would rub
but it sealed itself.
[img]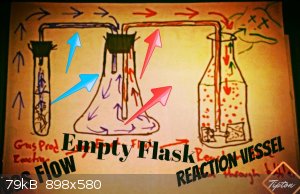 [/img] [/img]
|
|
|









