prole
Hazard to Self

Posts: 94
Registered: 4-8-2005
Member Is Offline
Mood: No Mood
|
|
Switching rcving flask in vacuum distillation
I'm getting ready for my first vacuum distillation. I'm unsure how to change the receiving flask when necessary. Will removing it while
vacuum and heat are on give me problems? Or should I remove heat, then disconnect vacuum before changing flasks? To my primitive mind, the latter
seems more logical, but really time-consuming (I'm using an oil bath). The former seems to me that the sudden return to 1 atm would cause all to
boil suddenly and ruin my day. I may have 2-3 fractions to collect, so I'd like to be armed with the right info before I proceed.
Before anyone gets pissed, I looked in all my reference books, I UedTFSE, and did a google search. I found lots of useful ideas, but this one was
never addressed. Thanks for the help.
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
Crashing to 1atm is only a problem if it's at <B>higher</B> pressure, so that the temperature is raised above the 1atm boiling point
and thus your experiment ends up on the ceiling (or worse in the case of steam boilers!).
I don't know of any problem with letting the vacuum out (), unless
atmospheric contamination is a problem. In which case, you'd just backfill with inert gas...
Couldn't you fit a manifold with stopcocks to change the recievers without dismantling it? I imagine that would be the professional way to do
it.
Tim
|
|
|
vulture
Forum Gatekeeper
Posts: 3330
Registered: 25-5-2002
Location: France
Member Is Offline
Mood: No Mood
|
|
Crashing to 1atm during distilling is suicide, not only for your glass, but also for you! This is an extreme hazard when distilling flammable
substances.
You should consider buying a spider.
One shouldn't accept or resort to the mutilation of science to appease the mentally impaired.
|
|
|
prole
Hazard to Self
Posts: 94
Registered: 4-8-2005
Member Is Offline
Mood: No Mood
|
|
I envisioned a manifold-apparatus, along with a large price tag to fit 24/40 glass. I haven't come across any. I'm sure I could
'engineer' something from the hardware store, but I'd worry about losing vacuum from a faulty seal.
Atmospheric contamination is not a problem for my planned run, and flammable liquids will not be used. I am still confused with the outcome of
crashing to 1 atm. Vulture, do you think the sudden change in pressure would destroy my glass? I've read so many reports of folks switching
flasks to collect different fractions under vacuum, and this was NEVER mentioned! I'd practice with water, but not at the expense of a pricey
glass collection!
I'm not familiar with a spider. Is that a manifold, and is that the name one would search under if one desired to aquire one?
Thanks
-prole
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
Crashing to 1 atm can cause explosions, if distilling flammable liquids. You will find a spider under 'distilling receivers' in the
catalogues of lab-glass sellers. That's how it looks like:

I suppose a construction of a tap over a droping funnel with a tap at the end of a distilling apparatus could be a low cost alternative for a spider,
but I didn't have tried it out.
~Mephisto
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
You could use an equalizing addition funnel for your first receiver , the type having a stopcock also in the equalizing tube . Some of these will
have a three way stopcock in the equalizing tube which can be positioned to vent the lower portion to atmosphere or a side line .
If the stopcock is a simple straight style instead , then you will need a two neck
flask for the receiver below for equipping the second neck with stopcock equipped inlet adaptor . Here you can release the vacuum for removing the
receiver flask while not interrupting the vacuum or distillation in progress . This will allow changing receiver flasks or withdrawing samples
without interrupting the distillation .
A side takeoff tubulature straight vacuum adapter will be needed between the top of the equalizing addition funnel and the bottom of the condenser .
Here is where your vacuum source will attach . The addition funnel acts as a feed through when its lower valve is open , and as a temporary reservoir
when the valve is closed .
|
|
|
Mephisto
Chemicus Diabolicus
Posts: 295
Registered: 24-8-2002
Location: Germany
Member Is Offline
Mood: swinging
|
|
That's how I imagine a low cost distilling receiver for interruptless vacuum distilling.

~Mephisto
|
|
|
prole
Hazard to Self
Posts: 94
Registered: 4-8-2005
Member Is Offline
Mood: No Mood
|
|
Thanks for the helpful responses, everyone. Prole is blushing with embarrassment, because he never thought to try his pressure-equalization funnel
that he has lying around...
The spider looks very interesting, but I'd need to save more empties (10 cents in Michigan) to buy one. Prole's wife doesn't approve
of any more expensive additions to his collection, so he must improvise with what he's got.
|
|
|
runlabrun
Hazard to Others
Posts: 172
Registered: 4-12-2004
Member Is Offline
Mood: No Mood
|
|
if you are using a vacuum pump you would find it hard to pull apart the joint at the reciever anyway... with my pump running i cant take any joints
apart until i open the purge valve on the vacuum manifold, its just too damn hard.
Use the diagram mentioned above, its as close as your going to get to a home-made perkin reciever which is the pro fraction cutter apparatus used
where a rotator reciever is not appropriate.
-rlr
|
|
|
BromicAcid
International Hazard
Posts: 3246
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
I've had my share of bad experiences disconnecting a flask under vacuum, it is a vacuum afterall, it sucks up things, like when I disconected my
distilling flask from a reduced pressure distillation and the liquid in the distilling flask suddenly pulled itself into the liebig condenser and into
the recieving flask.
Of course slowly increasing the pressure with some kind of gas letoff is advisible. However for changing the recieving flask you don't want to
depressurize and pressurize over and over. Instead the 'spider' shown above is a good alternative. Alternatively in my school it is called
a 'cow' and the different slots for different flasks are called 'udders' 
[Edited on 8/9/2005 by BromicAcid]
|
|
|
12AX7
Post Harlot
Posts: 4803
Registered: 8-3-2005
Location: oscillating
Member Is Offline
Mood: informative
|
|
Sure you aren't in Wisconsin, Bromey? 
Tim
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
I'm impressed that they even did a distillation warranting the use of one in Bromic's high school. Unless you meant school as in
college\university. Either way, we never touched any 24/40 glassware (or any jointed glass) in high school chemistry.
|
|
|
BromicAcid
International Hazard
Posts: 3246
Registered: 13-7-2003
Location: Wisconsin
Member Is Offline
Mood: Rock n' Roll
|
|
Yeah, I ment college, and it wasn't until my fourth year that we used anything 24/40, let alone the 'cow' 
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
Good, I was getting very envious of your high school's science curriculum. :-p
|
|
|
bio2
Hazard to Others
Posts: 447
Registered: 15-1-2005
Member Is Offline
Mood: No Mood
|
|
changing flasks under vacuum
This is how you do it with nothing special only due caution. Maybe you should practice with water if this doesn't seem obvious.
When the fraction coming over is finished and the next starts to climb the column quickly slow the stirring way down turn off the heat and bleed in a
little air at the valve on the pressure manifold. Then valve off the system and leave the pump running. Wait a few minutes for the temp to drop then
slowly open the bleed valve letting the system reach atmospheric. QUICKLY change recievers, this avoids superheating. Now slowly reapply the vacuum
and the boiling can be controlled by varying the pressure and stirring speed. Turn the heat back on and and ramp it up slowly and if bumping becomes
to bad at first make the adjustments until boiling smoothly.
Not hard as it sounds and takes about 10-20 minutes total before the next fraction starts to collect. A refrigeration manifold works well just please
don't try gradually pulling the tubing off the vacuum adaptor nipple. It may work most of the time but one day you will be very sorry that you
did this.
Man, I must be getting old! In 10th grade the chemistry teacher let me order my chems for the next year and in the 9th grade we had all 24/40
glassware with 250ml as the smallest flasks. I hate this microshit! We were azeotropically distilling esters we synthesized also in the 10th grade.
Seems that a lot of kids are dumbed down these days when it comes to science in general.
[Edited on 15-8-2005 by bio2]
|
|
|
Chris The Great
Hazard to Others
Posts: 463
Registered: 29-10-2004
Location: Canada
Member Is Offline
Mood: No Mood
|
|
This little improvished device comes from an article from rhodium titled 'Simple glass to fancy glass' written by MaDMAx.
Basically, when the fraction begins to change, you close the valve letting the distillate enter the reciever. The reciever is then brought to
atmospheric pressure using the three way stopcock, disconnected, and then a new flask or whatever is attached. The vacuum is then slowly reapplied by
slowly turning the three way stopcock back. Once the flask is under vacuum, the funnel stopcok is opened and the fraction is collected. Repeat as
necessary.

|
|
|
bio2
Hazard to Others
Posts: 447
Registered: 15-1-2005
Member Is Offline
Mood: No Mood
|
|
Yes, that is the most simple proper way to do it.
I would add however that a very fine needle valve for air bleed is still required at the pump.
Those 3 way stopcocks as depicted (plug type) offer almost no controlability as they are only 1/4 turn and are basically on or off.. Anyone who has
ever used them can attest to that.
|
|
|
EmmisonJ
Hazard to Self
Posts: 89
Registered: 5-1-2009
Member Is Offline
Mood: No Mood
|
|
what about a 3-way adapter that goes in between the tubing from the vacuum adapter to the vacuum pump. one nozzle has non-collapsible (like
crosshatched) tubing that goes to the vacuum adapter, another nozzle has collapsible tubing that has a clamp holding it shut, and the last nozzle has
non-collapsible (like crosshatched) tubing that goes to the vacuum pump.
when you need to change flasks just very very slowly open the clamp to slowly bleed air in (basically introducing your own leak). once the clamp is
fully opened up and the tubing has allowed atmospheric pressure throughout the apparatus then leave the vacuum pump running and disconnect the tubing
from the vacuum pump. quickly change your flasks to avoid superheating and reconnect the tubing to the vacuum pump then slowly clamp the collapsible
tubing again until it's tight and your apparatus is in vacuum again.
wouldn't that work? it'd only require a quick run to your local hardware store. see picture
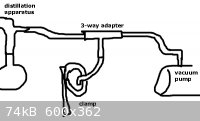
[Edited on 6-11-2009 by EmmisonJ]
|
|
|
aonomus
Hazard to Others
Posts: 361
Registered: 18-10-2009
Location: Toronto, Canada
Member Is Offline
Mood: Refluxing
|
|
It wouldn't be too hard to make a small copper manifold from plumbing parts and compression fittings and needle valves to make a 3 way adapter with a
vacuum gauge and insert it inline... whether it has a good enough seal is another thing.
I'd think that once your vapor temperature starts to rise, you would remove the heat and bleed the vacuum just a tiny bit so that it stops rising,
allow it to cool a bit, then slowly bring it back up to 1atm. Switch flasks, close system, and slowly depressurize it until its open to vacuum and
then begin stirring/heating. Copper fittings and time are cheaper than the glassware for the apparatus shown by Rhodium.
That being said, the apparatus with the non-pressure equalized addition funnel would make it fast and easy to switch out without bringing the whole
system up and down from vacuum.
|
|
|
Contrabasso
Hazard to Others
Posts: 277
Registered: 2-4-2008
Member Is Offline
Mood: No Mood
|
|
ebay has the proper tool! Item number: 110389116798 & Item number: 120419493442
Keep searching "Quickfit pig" You fit three collection flasks, pump down and distil then when one distillate is complete you turn (greased cone
please!) the pig so that the next receiver is in line. Baffcat is a good guy, I've dealt with him, but you may prefer to watch ebay in your area to
minimise shipping costs
|
|
|
anotheronebitesthedust
Hazard to Others
Posts: 189
Registered: 24-6-2007
Member Is Offline
Mood: No Mood
|
|
What is wrong with turning the vacuum pump off? Are flasks going to implode/explode? Or is it just wear and tear on the pump? Or laziness in not
wanting to wait for the vacuum to build again?
|
|
|
watson.fawkes
International Hazard
Posts: 2793
Registered: 16-8-2008
Member Is Offline
Mood: No Mood
|
|
It's not just waiting for vacuum to build,
it's waiting to reestablish steady-state conditions in a fractionating column. It works, but it's mighty, mighty slow.
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
Quote: Originally posted by anotheronebitesthedust  | | What is wrong with turning the vacuum pump off? Are flasks going to implode/explode? Or is it just wear and tear on the pump? Or laziness in not
wanting to wait for the vacuum to build again? |
The contents of a hot vacuum distillation have been known to
explode if air is admitted to them while hot. Not often, but trust me, they have.
|
|
|
mr.crow
National Hazard
Posts: 884
Registered: 9-9-2009
Location: Canada
Member Is Offline
Mood: 0xFF
|
|
How well would the addition funnel stand up to vacuum?
A glass stopcock would be more airtight then teflon. You would only need a small one, like 60mL to catch the distillate until a new flask is attached.
If you only need 2 fractions you could use just a pressure equalized addition funnel and a flask. When you are done with the first fraction just turn
off the stopcock.
|
|
|













