Phthalic anhydride by thermal decomposition of phthalic acid (Photo-essay)
Preparation of phthalic anhydride by the thermal dehydration of phthalic acid
(photo-essay)
by SplendidAcylation
References
[1] (Magpie's write-up)
[2] Systematic Organic Chemistry by Cumming, Hopper, and Wheeler (p.264)
[3] (video) ~25% yield
[4] (video) ~15% yield
[5] (video) no yield?
[6] (video) doesn't sublimate
Introduction
This project was inspired by Magpie's write-up[1], in addition to various videos on YouTube[3-6], with the ultimate desire to
obtain some phthalic anhydride for various experiments.
Being the kind of person who likes to make even the simplest reagents that many would simply purchase, I set out upon a mission to obtain phthalic
anhydride by the thermal dehydration route, from phthalic acid.
I initially attempted to prepare phthalic acid by extracting phthalate esters from PVC gloves, but this was fruitless, as it seems that the PVC gloves
I used were phthalate-free.
YouTube videos 3-5 use the traditional sublimation apparatus consisting of a beaker with an RBF full of cold-water on-top.
Although this technique is effective for many sublimation experiments, it is less than ideal for phthalic anhydride, as this compound tends to form
very light, woolly crystals, so the quantity of crystals that may be captured this way, before the beaker fills with crystals, is very small.
Magpie, and the author of video 6, do not recover the sublimated product, instead retaining the melted phthalic anhydride after the water has been
driven off, this is less tedious, but is prone to purity issues:
Magpie later reported[1] that phthalic anhydride prepared in this method is impure and contains unreacted phthalic acid, suggesting that
anyone attempting to repeat his procedure should recover the sublimated anhydride instead.
It is possible to separate phthalic anhydride from unreacted phthalic acid by extraction with chloroform, in which the anhydride is soluble, but the
acid isn't.
However, this approach isn't ideal, due to the toxicity of chloroform and the added complexity.
Properties of phthalic anhydride
Phthalic anhydride is a white solid, with a melting-point of 131.6°C and a boiling-point of 295°C, it has a tendency to sublimate, forming a mass of
long needle crystals, resembling cotton wool.
Etymology:
Shortened form of naphthalic, from naphthalene + -ic, from naphtha + -ene.
(ScienceMadness Wiki)
(Wikipedia)
Unlike intermolecular anhydrides, such as acetic anhydride, which tend to be much more reactive, reacting immediately with water and atmospheric
moisture, phthalic anhydride is relatively unreactive, even upon contact with water; It does, however, react slowly with boiling water, hydrating to
phthalic acid.
Phthalic anhydride is a versatile precursor to phthalic acid and its derivatives (esters, phthalimide, &c), in fact, phthalic acid is seldom
prepared in industry, its derivatives instead being prepared directly from the anhydride.
This is due to the efficient industrial routes to phthalic anhydride, namely, the oxidation of naphthalene or o-xylene.
However, a much more accessible route to phthalic anhydride available to the amateur chemist is the thermal dehydration of phthalic acid, which can be
prepared by the saponification of phthalate esters used as plasticisers in PVC:
PVC ----Extraction----> Phthalate esters ----Saponification----> Phthalate ion ----Acidification----> Phthalic acid ----Heating---->
Phthalic anhydride
For the following experiments, however, the phthalic acid used was prepared by the saponification of reagent-grade diethyl phthalate.
Reaction
The dehydration takes place above 210°C, with the phthalic acid melting and the phthalic anhydride and water escaping as a vapour, the anhydride
condensing in the form of long needle crystals.
The reaction for the thermal dehydration of phthalic acid is given below:
C6H4(COOH)2 → C6H4(CO)2O + H2O
----------------------------------------------------
......166.13................. 148.1.........18.02
Summary of following experiments
In Experiment 1, the traditional RBF+beaker approach was used, resulting in a percentage yield of 28.0%.
In Experiment 2, an alternative apparatus (p.31 of [2]) was used, resulting in an even lower percentage yield of 24.4%.
In Experiment 3, a better apparatus was then devised, employing a glass dish wherein the phthalic acid was placed inside a nickel crucible, induction
heated from outside (the apparatus is described in more detail below), this resulted in a percentage yield of 78.5%.
In Experiment 4, Experiment 3 was repeated on a larger scale, five times, resulting in an almost quantitative yield (94-99%)
Experiment 1 (1g scale):
1.00g (6.02mmol) of phthalic acid, previously prepared from diethyl phthalate, was placed in a 100mL Pyrex beaker on an asbestos gauze over a flame,
and a coarse metal gauze was placed over the beaker, with an inverted funnel on top of the gauze.
This apparatus was chosen because the gauze would pass the vapours but prevent the crystals from falling back into the beaker, and a funnel was used
to capture the crystals as it had a stem through which the steam could escape.
The beaker was heated, via the asbestos gauze, over the flame, whereupon the phthalic acid began to melt and bubble, with crystals forming on the
walls of the beaker, and liquid phthalic anhydride or phthalic acid refluxing.
Some needles of phthalic anhydride condensed on the metal gauze itself, with some woolly crystals condensing inside the funnel as intended, however,
significant quantities of phthalic anhydride vapour, and steam, were escaping due to the poor seal between the gauze and the beaker, so the gauze was
removed and the funnel was lowered such that it surrounded the top of the beaker.
Slowly, needles of PAN began to form in the funnel, however they frequently fell back into the beaker, so it was necessary to repeatedly discontinue
heating, carefully remove the funnel to avoid dropping the crystals in, remove the crystals, and return the funnel.
Each time this was done, some vapour escaped.
The crystals obtained seemed fairly pure, however they may have contained appreciable amounts of water, as there was no way for the steam to escape
except via the stem of the funnel.
The process was painstaking, eventually very little substance remained in the beaker, so the process was discontinued.
Yield: 0.25g (1.69mmol), this corresponds to a percentage yield of 28.0%.
Experiment 2 (4g scale):
A better design was devised, consisting of a small casserole dish inverted (with the lid on the bottom) and placed on the hot-plate.
This was inspired by a similar apparatus, consisting of two watch-glasses with a perforated filter-paper in between, described on p.31 of [2].
A sheet of printer paper was pierced with ~100 small (1mm) holes in a circle equal to the diameter of the dish, and the paper was sandwiched between
the lid and base of the dish.
Into the lid (which was at the bottom, sitting on the hot-plate) was placed 4.00g (24.1mmol) phthalic acid.
The hot-plate was turned on with the PID controller set to 255c, and a damp paper-towel was placed on the top of the dish to cool it somewhat.
Once the temperature exceeded ~200c, some phthalic anhydride vapour was visibly puffing through the holes in the paper.
Eventually, long needles began to form, surrounding the holes in the paper, the crystals slowly building up inside the dish.
This seemed promising, however, once the temperature rose further, an unforeseen problem arose;
The paper separator, warmed by the hot surface below, eventually became hot enough to melt the phthalic anhydride crystals, causing the needles to
melt as they fell down onto the paper.
This problem gradually worsened, with the paper eventually becoming soaked with melted phthalic anhydride.
A significant quantity of crystals remained un-melted as they were stuck to the glass, or nearer the edges of the paper where the temperature was
lower, however this was not ideal.
After heating for ~30 minutes, heating was discontinued, and the apparatus was allowed to cool, whereupon the crystals were scraped off of the glass,
however this yielded only ~0.25g of phthalic anhydride.
In order to recover the phthalic anhydride melted into the paper, the paper was cut up into pieces and it was attempted to re-sublimate the anhydride
directly from the paper, by placing the paper in the lid on the hot-plate, and replacing the paper separator with a new one.
This time the temperature was kept somewhat lower to prevent the same problem from recurring; This was somewhat successful, however the paper
gradually browned with heating, giving off some unpleasant fumes which inevitably diminished the purity of the product.
After most of the available sublimate appeared to have formed, the apparatus was again allowed to cool and the crystals collected, the browned pieces
of paper being discarded.
Yield: 0.87g (5.9mmol), this corresponds to a percentage yield of 24.4%.
The problem with this apparatus was that the temperature of the paper separator exceeded the melting-point of the anhydride.
In order to resolve this issue, a better apparatus needed to be devised.
An alternative apparatus was illustrated on p.32 of the aforementioned book ([2]); It consisted of an inverted watch-glass placed upon an asbestos
board having a small circular hole in the centre; A crucible was placed in the circular hole, and heated from below with a Bunsen burner.
In the book, this apparatus is recommended for substances that are difficult to sublimate, however it seemed that it would also solve some of the
problems with the apparatus used in Experiment 2, as the asbestos board would be unlikely to reach as high a temperature as the paper separator, given
that the heating would be localised to the crucible.
A modification of this apparatus was ultimately used, with a nickel crucible containing the phthalic acid being placed inside a large Pyrex casserole
dish, and heated from below using an induction heater (driven by the ZVS driver).
This apparatus was devised such that the sublimate would condense on the cold walls of the glass dish, and most of the crystals that fell down would
land on the glass base, which would be unheated; Only a small quantity would fall into the hot crucible (whose surface area was small compared to the
glass base of the dish), and the crystals that did fall into the crucible would re-sublimate.
Experiment 3 (induction heated) (1g scale):
1.00g (6.0mmol) phthalic acid was placed in the nickel crucible, which was placed in a large glass Pyrex casserole dish.
The induction heater was switched on; within a few minutes, the glass became clouded with condensation, and the white solid began to melt and bubble,
with woolly crystals depositing on the glass above the crucible.
Shortly thereafter, small crystals began to swirl around inside the dish, eventually coalescing as woolly crystals which gradually filled the dish.
After a total of approximately five minutes, the crucible was empty, with a very small quantity of tarry residue remaining therein, the dish filled
with woolly crystals.
The induction heater was switched off and the apparatus was allowed to cool down, whereupon the dish was opened, revealing a large mass of light
woolly needle crystals filling the shape of the dish, these were readily collected into a wad of wool.
Significant quantities of fine white powder were deposited on the glass, quite different in form than the needle crystals.
Some of the product had melted onto the glass dish in a circle around the crucible, presumably due to radiation from the crucible, in addition to heat
conducted through the glass from the work coil of the induction heater (which was not water-cooled, so it reached a fairly high temperature due to
resistive heating).
All of the product (the woolly crystals, fine white powder, and melted material) was returned to the crucible and re-sublimated, exactly in the same
manner as before.
During the re-sublimation, the solids melted and began to bubble, however no condensation formed on the glass, because there was no reaction taking
place that released water, so it was easier to see what was happening in the dish.
In this case, much less of the product deposited as a fine powder, with the majority forming woolly crystals, this is suspected to be due to the lack
of water, compared with the initial reaction; perhaps the water caused some of the crystals to stick to the glass and form improperly?
Alternatively, perhaps the re-sublimation yielded a purer product.
After the second sublimation, the experiment yielded 0.70g (4.7mmol) of woolly needle crystals of phthalic anhydride, this corresponded to a
percentage yield of 78.5%, around three times higher than the yield from Experiments 1 & 2.
Experiment 4 (induction heated) (4g scale):
The same procedure, used in Experiment 3, was followed, this time using 4.00g (24.1mmol) of phthalic acid.
Figure 1: 4.00g phthalic acid in nickel crucible:

Figure 2: Crucible containing phthalic acid inside dish, with induction heater work-coil visible:

When the induction heater was switched on, the phthalic acid would first melt before sublimating.
Figure 3: Molten phthalic acid, sublimating:

The glass would become fogged with water vapour, obscuring the view inside.
Figure 4: The sublimation in progress, with condensation obscuring the view inside the dish:

A short while later, vapours would be seen swirling around inside the dish, with a circle of white crystals on the top of the dish above the crucible.
Then woolly crystals would gradually become visible inside the dish, some of which would fall back into the crucible and re-sublimate from the
crucible in a puff of vapour.
Eventually, the dish would be full of light woolly iridescent needle crystals, the contents of the crucible would be liquid and bubbling by this
point.
The end-point would be indicated by the observation that the crucible was empty.
The sublimation took only ~5 minutes for each 4g run.
Figure 5: The mass of woolly crystals after the reaction was complete:

In each attempt, the sublimate crystallized both as wool and as white powder stuck to the glass.
The wool was loose and easily gathered, while the white powder had to be scraped from the glass with a blade.
The powder was much denser, a thin layer of the powder weighing more than volumes of wool.
Figure 6: The woolly crystals gathered up into a wad, with the fine powdery crystals and melted solids visible:

The product was not re-sublimated, instead, it was decided to retain the wool, powdery crystals, and melted product, separately, until a number of
runs had been carried out, and then re-sublimate it all at once.
The yield of phthalic anhydride after the first thermal decomposition/sublimation (no re-sublimation, as mentioned above) was 3.37g (22.8mmol), this
corresponded to a percentage yield of 94.5%.
Since 4g seemed to be a sufficient quantity of phthalic acid to fit into the crucible (it filled it to around half-way), and given that the yield was
almost quantitative, the procedure was repeated four more times, with 4g of phthalic acid each time, in order to obtain a sufficient quantity of
phthalic anhydride for future experiments.
All of the data is tabulated below:
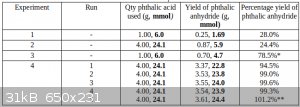
*In this experiment, a re-sublimation was carried out prior to weighing the product, so this figure is probably somewhat lower than it should have
been, compared with the other data, all of which represent the yield without a re-sublimation.
**In the fifth and final run in Experiment 4, the yield was slightly higher due to more thorough scraping of the powdery sublimate from the glass, the
impossibly high yield being due to product from the previous sublimations.
Usually, around 1.45g of the product was collected as wool, the remaining ~2g being in the form of fine powder or melted solids.
In the fifth run, due to a problem with the induction heater, the process was stopped half-way and the wool was gathered into a clump before resuming
the process.
Although the final yield was much the same, a much larger proportion of the product (2.45g compared with the usual 1.45g) was in the form of wool,
with much less in the form of the denser powder.
The reason for this is uncertain, perhaps the glass was not allowed to become excessively hot, therefore less product melted onto the glass.
In total, 19.31g of phthalic anhydride, collected as wool and as fine powder and melted solids, was obtained.
Figure 7: 19.31g crude phthalic anhydride, as woolly crystals, fine powder, and fused lumps:

This entire quantity was re-sublimated, using the same procedure as before (using the induction-heated nickel crucible inside the glass dish).
Figure 8: 4.00g crude phthalic anhydride in crucible, for resublimation:

Figure 9: The resublimation in progress, with the woolly crystals clearly visible due to the lack of condensed water obscuring the view:

It was necessary to carry out the re-sublimation in 4g batches due to the small volume of the crucible, furthermore, numerous re-sublimations were
necessary, each time collecting the wool and returning the powdery sublimate and melted solids to the crucible.
This was done because it was desired to obtain all of the product in the form of wool, although this was probably unnecessary.
Eventually, 18.59g of wool was obtained, in addition to 0.14g of fine powder scraped from the glass after the final re-sublimation.
Final yield: 18.73g (126.5mmol), 97.0% of the mass of the crude product prior to re-sublimation.
An attempt at melting-point determination was made; A sample of the product was placed in a small glass tube (I.D. = 1mm) sealed at one end, which was
secured using an elastic band to a K-type thermocouple, both of which were immersed in mineral oil, the oil was magnetically stirred and pre-heated to
100°C, whereupon the temperature was increased slowly thereafter.
The sample began to change in appearance at 126°C, the first signs of liquid were visible at 129°C, it was almost fully liquid by 131°C, and fully
melted by 132°C.
This corresponds nicely with the published melting-point of 131.6°C.
Conclusions:
The induction-heated approach was far superior compared with the traditional beaker + RBF approach, however the induction-heating was not strictly
necessary, an alternative approach would be to use small electric heating-elements.
The apparatus would work even better if a larger glass (or other material) dish were used, this would allow more space for the woolly crystals,
perhaps reducing the fraction that deposited on the glass as a fine powder.
Perhaps a large desiccator would be a more well-suited apparatus.
When this procedure is documented in the literature, no mention is ever made of what happens to the water vapour formed in the reaction, and whether
it is necessary to dry the phthalic anhydride afterwards, perhaps in a desiccator.
It is assumed that the water vapour readily escapes once the dish is opened, or perhaps it remains in the gas-phase while the surrounding dish (or
watch-glass or beaker, whatever vessel is used) remains warm.
Either way, it is never suggested in the literature to dry the anhydride to remove traces of condensed water vapour from the reaction, so it is
assumed that the product will be sufficiently pure without drying.
It would probably not be advisable to dry the phthalic anhydride by heating in moist ambient air, as this may cause some of the phthalic anhydride to
hydrate, re-forming phthalic acid, so, if it were necessary to ensure that every trace of water were removed, perhaps vacuum-desiccation would be the
best approach.
In these experiments, the product was re-sublimated for three reasons:
1. In order to remove any traces of water that may have been present.
2. To ensure that the product was as pure as possible (as it was suspected, probably erroneously, that the white powder stuck to the sides of the
dish might have been un-reacted phthalic acid).
3. To obtain the entirety of the product in the form of woolly crystals (again, due to the probably erroneous assumption that the other physical
forms may have been less pure).
The process of re-sublimation added complexity to the process, especially the iterative process of repeated re-sublimation to obtain the entirety of
the product in the woolly form; Since this process was probably unnecessary, it could be forgone in future attempts, rendering the procedure very
facile.
I have attached two versions of the write-up (one with images and one without), in both OpenDocument and PDF format.
Furthermore, I have attached my write-up for the preparation of the phthalic acid by the saponification of diethyl phthalate.
There is also a short (65s) video showing the resublimation at 10x speed here.
Thanks for reading! Please let me know if I have made any mistakes, or if there is anything you think I have forgotten to include, etc. 
The OpenDocument version of the write-up with images is not yet available, as it is too big at the moment (25MB) so I might have to reduce the image
quality.
Attachment: Phthalic acid.odt (103kB)
This file has been downloaded 237 times
Attachment: Phthalic acid.pdf (85kB)
This file has been downloaded 258 times
Attachment: Phthalic anhydride (without images).odt (54kB)
This file has been downloaded 231 times
Attachment: Phthalic anhydride (without images).pdf (63kB)
This file has been downloaded 236 times
Attachment: Phthalic anhydride (including images).pdf (2.4MB)
This file has been downloaded 274 times
[Edited on 4-1-2023 by SplendidAcylation]
|












