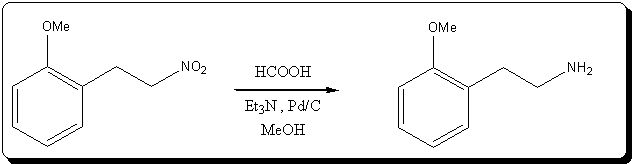
This three-phase system consists of an aqueous formate salt solution, an organic, substantially water-immiscible, solution containing the substrate
which is to be reduced and a third solid phase of a Group VIII metal supported catalyst, without the need of using a solvent, high temperatures or a
phase transfer catalyst.
[...]
A major advantage of the heterogeneous catalyst systems over the homogeneous ones lies in the ease with which the catalyst can be separated after
reaction by simple filtration and reused afterwards. |

 ). Peer-reviewed research chemists can be wrong. Patent chemists can be wrong, and even prone to mislead.
). Peer-reviewed research chemists can be wrong. Patent chemists can be wrong, and even prone to mislead.







 ), so it was not triethylammonium
chloride (mp= ~240°C) [Note 5]
), so it was not triethylammonium
chloride (mp= ~240°C) [Note 5]