Crystalline hydrogen sulfide-formaldehyde reaction products known as
formthionals are also obtained by saturating 37 per cent formaldehyde with
hydrogen sulfide at temperatures in the neighborhood of 40 to 50°C.
Heat is evolved by the reaction and product is precipitated, almost completely
solidifying the reaction mixture. A product of this type obtained
from neutral formaldehyde melted at 80°C after crystallization from chloroform
and contained 51.5 per cent sulfur. When pure, these compounds
are colorless and have little odor.
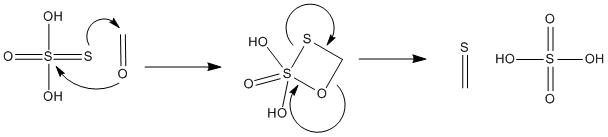
Under strongly acidic conditions, trithiane or trithioformaldehyde,Sulfuretted FORMALDEHYDE
methylene glycol derivatives of the type described above are converted to
trithiane on heating with concentrated hydrochloric acid. Trithiane is
also produced by reaction of 37 per cent formaldehyde, sodium thiosulfate,
and hydrochloric acid. |



 ) was dissolved into the formaldehyde sol.
(at ca. 10c). The HCl was then added, the solution slowly precipitated very fine crystals over the course of a few minutes. The solution was vac
filtered, yielding beautiful tiny light blue needles (filtrate boiled in filter flask durring vac filtration...isocyanic acid?). The filter cake did
not readily dry by passing air through it at the pump, so it was washed with ~2mL of isopropanol (if only I had it on hand I would have used ether).
The isopropanol unfortunately dissolved ca 50% of the product leaving a nearly white non-crystalline (odd!) powder.
) was dissolved into the formaldehyde sol.
(at ca. 10c). The HCl was then added, the solution slowly precipitated very fine crystals over the course of a few minutes. The solution was vac
filtered, yielding beautiful tiny light blue needles (filtrate boiled in filter flask durring vac filtration...isocyanic acid?). The filter cake did
not readily dry by passing air through it at the pump, so it was washed with ~2mL of isopropanol (if only I had it on hand I would have used ether).
The isopropanol unfortunately dissolved ca 50% of the product leaving a nearly white non-crystalline (odd!) powder. 












