Boffis
International Hazard

Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
The chemistry of Sorbic acid
Two threads have appeared recently on the esterification of sorbic acid (1,2) and previously I have posted questions about possible Diels Alder type
condensations of sorbic acid (3). These threads apart I would like this thread to become the resting place for all things sorbic.
Sorbic acid is an interesting substance having two adjacent double bonds, one of which is adjacent to a carboxylic acid group and this affects its
reactivity. I have already experimented with adding bromine across the double bonds in a chlorinated solvent and I am currently experimenting with
adding chlorine in an aqueous medium. Being a conjugate diene sorbic acid readily undergoes Diels Alder type addition reactions (e.g. (4), but
numerous other references exist)
There are numerous papers out there concerned with the reaction of sorbic acid or sorbate ions in food with other food component and preservatives
such as sulphite (7), thiols (7), nitrite (7,8,9,10) and ammonia(7). While these are mostly dealing with very dilute solutions they point to some
interesting chemistry that I feel is worth following up at preparative concentrations as sorbic acid or its potassium salt are so readily available
and cheap.
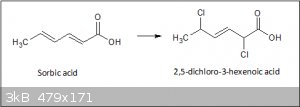
The reaction I alluded to above concerning the addition of chlorine to sorbic acid in aqueous solution is based on a method of analysing sorbates in
food (5). The fact that the reaction is practically quantitative and carried out using very simple, OTC chemicals, makes it attractive. Initial
results look promising, the resulting compound is claimed to be 2,5-dichloro-3-hexenoic acid. The 2,5-addition with migration of the remaining double
bond to the 3 position seems to be a common theme in aqueous solutions of sorbic acid (7). The rapid addition of bromine in carbon tetrachloride is
supposed to be 4,5 addition followed more slowly and with forcing conditions the 2,3 positions, ultimately giving hard glassy crystals of
2,3,4,5-tetrabromohexanoic acid.
The reaction of sulphite ions and thiols with sorbic acid give rather unstable addition products that tend to undergo reversible condensations (7).
Nitrite ions or perhaps nitrous acid react with sorbic acid in a complex fashion producing an array of weird compounds such as ethyl nitrolic acid, a
furoxan derivative and a dinitropyrrole. These reactions tend to have been investigated in rather dilute solutions by food scientists but may still
merit further investigation as the reagents are cheap and available.
I found on paper that claims that the oxidation of sorbic acid with dichromate and acid yields malondialdehyde (11). The reaction of the latter with
thiobarbituric acid produces a red polymethine dye that is the basis of the photometric measurement for sorbic acid. However, there is no explanation
of the mechanism of malondialdehyde formation by this route and I must admit that I find it hard to believe. I have been unable to track down other
references to this method or the underlying reaction.
The references below are only a few of the considerable number of papers I have tracked down related to the chemistry of sorbic acid and potassium
sorbate. The Diels Alder addition type reaction seems a particularly fruitful avenue for amateur chemists.
1) Sorbic acid esters: http://www.sciencemadness.org/talk/viewthread.php?tid=155312
2) Synthesis of methyl sorbate: http://www.sciencemadness.org/talk/viewthread.php?tid=155365...
3) Diels Alder reactions of sorbic acid http://www.sciencemadness.org/talk/viewthread.php?tid=154454...
4) Craig & Shipman; 1952; Maleic anhydride adducts of sorbic acid and methyl sorbate; JACS; v74; p2905.
5) Spacu & Dumitrescu; 1967; Determination of sorbic acid with sodium chlorite, Talanta, v14, p981.
6) Farmer & Healey; 1927; Properties of conjugate diene compounds Part II Addition to Butadiene Esters; JCS p1060.
7) Khandelwal & Wedzicha; 1990; Nucleophilic reactions of sorbic acid; JFoodAdditives&Contam; v7; p685.
8) Namiki & Kada; 1975; Formation of ethylnitrolic acid by the reaction of sorbic acid with sodium nitrite; Agri. & Biol. Chem.; V39;
p1335-1336.
9) Kito et al; 1978; A new N-nitropyrrole, 1,4-Dinitro-2-methylpyrrole; Tetrahedron; v34, p505-508.
10) Osawa et al; 1979; A new furoxan derivative and its precursors formed by the reaction of sorbic acid with sodium nitrite; Tetrahedron Letts;
No.45; p4399-4402.
11) Molina et al; 1999; Spectrophotometric flow-injection method for determining sorbic acid in wines; LRA; v11, 299-303.
|
|
|
Syn the Sizer
National Hazard
Posts: 600
Registered: 12-11-2019
Location: Canada
Member Is Offline
|
|
Very interesting, I will be checking out the references. I am honoured to be referenced as well.
|
|
|
wg48temp9
National Hazard
Posts: 784
Registered: 30-12-2018
Location: not so United Kingdom
Member Is Offline
|
|
ethylnitrolic acid
From one of the OP's references is the facile synthesis of the interesting compound ethylnitrolic acid
"A solution of
sodium nitrite was added to a partially suspended solution of sorbic acid, each 0.5 M in
distilled water, at room temperature and heated
in a water bath at 90°C for 1 hr. The mixture colored yellow instantaneously, it turned
red and finally became dark red with heating,
where pH of the mixture changed from 4.3 at
the beginning to 6.0 in the final stage. The
reaction mixture was extracted two times with
dichloromethane and then three times with
ether, and each of the extracts was concentrated
in vacuo to give a dark red oily product."
From
Attachment: Formation of Ethylnitrolic Acid by the Reaction of Sorbic Acid with Sodium Nitrite.pdf (349kB)
This file has been downloaded 473 times
I am wg48 but not on my usual pc hence the temp handle.
Thank goodness for Fleming and the fungi.
Old codger' lives matters, wear a mask and help save them.
Be aware of demagoguery, keep your frontal lobes fully engaged.
I don't know who invented mRNA vaccines but they should get a fancy medal and I hope they made a shed load of money from it.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Aqueous Chlorination of Sorbic Acid
Enough of the intro stuff, now for some real chemistry.
Attempts at chlorinating Sorbic Acid with Sodium Chlorite
These experiments were developed from a paper by Spacu and Dumitrescu (1) on the determination of sorbic acid in foods by chlorinating the acid with
excess sodium chlorite and then measuring the excess chlorine in the reaction mixture. Sodium chlorite is a fairly accessible bleaching /sterilizing
agent and can sometimes be purchased on line from various suppliers. It comes as a white crystalline powder that consists of about 80% NaClO2 with the
remainder being mainly NaCl (salt).
Sodium chlorite reacts with excess hydrochloric acid to give sodium chloride and chlorine, approximately according to the equation:
NaClO2 + 4HCl -> NaCl + 4Cl + 2H2O
The reaction with sorbic acid is, according to Spacu and Dumitrescu:
NaClO2 + 4HCl + 2C6H8O2 -> NaCl + 2C6H8O2Cl2 +
2H2O
In the original work they were looking at low concentrations and higher concentrations do not always favour the same reaction route, however, in order
to make recovery of the resulting compound easier I used fairly concentrated solutions. One problem was that I had no data of the properties, such as
melting point and solubility, of the target compound so the first few experiments were carried out very much in the dark.
Experimental
The first 4 experiments were carried out on the same basic scheme:
Approximately 4g of sodium chlorite (80%) and 10g of potassium sorbate were dissolved in 40ml of water. In a separate beaker 23ml of 30% hydrochloric
acid were diluted with 50ml of cold water. The mixed salt solution was then added dropwise into the rapidly stirred acid over about 10 to 20 minutes.
The addition was accompanied but a slightly exothermic reaction and the mixture turns slightly yellow though this colour fades after 10 to 20 minutes.
Faster addition causes a slightly more pronounced exotherm and some chlorine is lost from the solution.
In Experiment 1 the beaker was stood in a water bath at room temperature, about 16 C, as soon as the exothermic reaction was noted. When the addition
was complete and the colour discharged the precipitated solid was filtered off, washed with a little water and dried to give 2.295g. The white
compound can be recrystallized from dilute ethanol, azeotropic isopropanol or water. The latter solvent proved the best but about 30ml per g are
required. When alcohols are used for the recrystallization the product was sticky.
After the filtrate had been discarded the product was identified as un-reacted sorbic acid.
Experiment 2 was conducted in exactly the same way except than the water bath was omitted. The ppt was filtered off as before (1.405g of sticky
unreacted sorbic acid) and the filtrate extracted with 40ml of ether and then two further portions of 30ml each. The extracts were combined and the
ether recovered by distillation to give a small volume of colourless liquid that soon crystallised into small rosettes of blades, these were filtered
from the small volume of slightly thick aqueous liquid; yield was 2.741g after drying. This material is quite distinct form sorbic acid being highly
soluble in water.
Experiment 3
In order to reduce the amount of unreacted sorbic acid this experiment was conducted as in Exp. 2 but with 4.616g of sodium chlorite. Unfortunately
this generated a stronger exotherm (circa 40° C) and the ppt became very sticky, it stuck to the stir bar, the beaker walls and the anything else it
touched. The sticky white material was ether soluble so the entire mixture was extracted with 50ml of ether in the beaker before pouring it into the
separating funnel. It was then extracted with two further 30ml portions of ether. The ether was gently distilled from the combined extracts and the
very pale yellow oily residue poured into a bowl and allowed to cool and the last of the ether to evaporate. About 2.5-3g of sticky white crystals
remained after a few hours. Recrystallization from azeotropic isopropanol (87%) gave 0.822g of slightly sticky unreacted sorbic acid. Foolishly I
rejected the filtrate; this would have been better evaporated and leached with water.
Initial Observation:
1) Higher reaction temperatures favour increased consumption of sorbic acid.
2) Higher reaction temperatures increase the amount of sticky polymer like by-product.
From these observations the obvious next step is to try a lower temperature reaction.
Experiment 4 was run with the 3.830g of sodium chlorite and 10.005g of potassium sorbate dissolved in 40ml of water and then chilled in the fridge to
about 4°. 23ml of 30% hydrochloric acid were diluted with 50ml of water and placed in the freezer until it had reached -16° C.
The acid was placed on the stirrer plate and stirred rapidly as the mixed salt solution was added. A dense white ppt formed that eventually stopped
the stir bar from moving; this is almost certainly simply precipitated sorbic acid. The temperature on completion of the addition was -5° so little
or no exothermic reaction had occurred. In fact no reaction apart from the liberation of free sorbic appeared to have taken place so the mixture was
left to stand and warm up slowly to room temperature overnight. The following morning the slurry was less thick and was easily vacuum filtered to give
3.800g of washed and dried sorbic acid. The yellow filtrate was extracted with three x 50ml portions of dichloromethane (DCM) and then the combine
extracts dried with magnesium sulphate. The DCM was distilled off until only about 10-15ml remained in the flask; this was poured out into a bowl and
allowed to evaporate slowly. No crystals formed but eventually a small volume of bright yellow, pleasantly ethereal smelling liquid remained. The
smell of the liquid is reminiscent of perchloroethylene and I suspect that decarboxylation has occurred to give a polychloro pentane or pentene.
Conclusions
The desired reaction product appears to be exceptionally soluble in water and this may account for the low recovery of the material in Exp.2,
basically a poor partition coefficient between water and ether. If this is the case, five or six extractions with ether may be required and possibly
the addition of KCl to assist extraction.
The optimum temperature is probably about 15-20° C since higher temperatures favour the formation of a sticky by-product. Lower temperatures involve
longer reaction times and this seems to result in a completely different reaction path.
A slightly larger excess of sodium chlorite coupled with a cooler temperature may reduce losses through unreacted sorbic acid and a greater number of
ether extractions cycles may improve the yield of product.
These findings point to my next set of experiments but I also intend to investigate the direction of mixing with an experiment where the acid is added
to the mixed salt solution.
1) Spacu & Dumitrescu; 1967; Determination of sorbic acid with sodium chlorite, Talanta, v14, p981.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Does any one have access to Reaxys or scifinder? I can't find any useful data on 2,5-dichloro-3-hexenoic acid and I would greatly appreciate a bit of
help here with say melting point, solubilty and reactions in order to determine the possible identity of my colourless crystals.
I have run some more experiments now, essentially the same as those described above but with varying excesses of HCl and chlorite but most importantly
temperature control. I have manage to get the yield of the ether extract up to 7.5g and the level of unreacted sorbic acid down to a small amount
though how small is difficult to tell since a viscous oil forms that sticks to the stir bar and the beaker and any remaining sorbic acid is
incorporated into this. The optimum temperature to maximise the yield of the water soluble acid is about 20 C but at this temperature the reaction
take several hour to lose its yellow colour and this favours the formation of by products. Lower temperatures (<5 C) favour the formation of the
heavy oil (S.G. about 1.4).
Out of curiousity I decided to investigated the chemical properties of the cream coloured stick oil that formed in some of these experiments. The oil
is completely soluble in both ether and isopropanol. An isopropanol solution of about 2.5-3g was mixed with a strong solution of sodium nitrite, 4g on
10ml of water. To my surprise there was no precipitate formed and no obvious reaction. On gentle warming a reaction began, the solution turned golden
yellow and an odourless gas was evolved (CO2?). After about 20 minutes at 40-45 C a pale brownish ppt had formed. I filtered this off and then
continued to heat the mixture a little more strongly. The IPA began to evaporate and more, slightly darker ppt formed from the increasingly dark
solution. I let the mixture cool and partially evaporate in a shallow bowl. When cold long slender colourless prisms had formed in amongst a
flocculant ppt. The mixture was warmed and a little water added to dissolve the crystals, the pale ppt filtered off and combined with the earlier
crop. The filtrate was allowed to crystallise and the cream coloured crystals recovered.
The filtrate was warmed again in a shallow bowl to assist evaporation. The colour darkened further, more gas was evolved and more ppt formed. The
volume of solution was now <10ml, it was filtered hot to remove the small amount of light brown ppt and cooled. An abundance of brown crystals
formed, obviously colourless but stained by residual liquor, 1.95g of slightly damp crystal after drying. These are awaiting further investigation.
The filtrate was acidified with dilute HCl (2M) but a mass of brown tar formed leaving and almost clear solution. The tar is insolulbe in ether or
IPA. The solutions were tested at intervals and remained almost neutral through out. Both the crystals and the light brown ppt are mildly energetic
and deflagrate vigorously when heated on foil.
My theory is that the cream coloured viscous oil is a mixture of unreacted sorbic acid and various chlorinated products that react with sodium nitrite
causing decarboxylation and nitration, ether via a nitroso intermediate or via halogen replacement.
I tried preparing a copper salt from the crystals, the grass green ppt was not energetic but swelled to a foam like mass much like mercury
thiocyanate.
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
I checked Reaxys for 2,5-dichloro-3-hexenoic acid. Believe it or not, I wasn't able to find a single reference for this compound! (The Spacu and
Dumitrescu paper you mentioned is not in the database of syntheses, apparently.) I was also unable to find any reactions of sorbic acid with either
sodium chlorite, chlorine dioxide, or chlorous acid in the literature. I was however able to find two papers giving the reaction of aqueous
chlorine/hypochlorous acid and sorbic acid:
https://sci-hub.tw/https://pubs.rsc.org/en/content/articlela...
https://sci-hub.tw/https://pubs.rsc.org/en/content/articlela...
Both papers suggest a series of chlorinated products are the outcome of the reaction, with hydrolysis to the alcohol and lactone formation being
observed to a large degree.
I also find it somewhat interesting that the authors seem to believe that the reaction of chlorite ion with hydrochloric acid produces chlorine, when
as far as I know the major product is chlorine dioxide. Maybe this would explain the lack of literature precedent.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Cryolite; those are two really interesting papers. They highlight the difference between the electrophilic attack of halogen which seem to give
rise to 3,4 substitution in sorbic acid as against the nucleophilic reagents which according to some of the papers I referenced above give mainly 1,4
substitution. This raises the issue of what the actual reactive species is in aqueous HCl/chlorite mixtures. I was also interested in the difference
the presence of water makes. I am just writing up the bromination of sorbic acid to both 4,5-dibromo-3-hexenoic and 2,3,4,5-tetrabromohexanoic acid
for this thread. These were carried out in essential dry conditions in DCM and carbon tetrachloride respectively and the results match the published
products. The chlorination in aqueous condition therefore may give entirely different products.
Incidentially, some years ago I did some experiments with caffeine. When dissolved in dilute HCl both sodium chlorite and sodium chlorate yield first
of all a well crystallised chlorocaffeine and then proceed to degrade the 5-membered ring to give dimethylalloxan and methylurea. In the first stage
of this reaction both sodium chlorite and chlorate seem to act as proxys for Cl2.
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Glad my search was of use to you-- let me know if you'd like me to search Reaxys for anything else.
I would definitely believe that the anhydrous halogenation gives the desired 2,5-dibromo and 2,3,4,5-tetrabromo products, as that would proceed by the
standard 1,4 addition reaction standard to dienes. For the aqueous chlorination, my hypothesis is that the halonium ion forms as normal but the
nucleophillic carbonyl attacks this ion as it forms and gives a lactone (which then may hydrolyze to the alcohol). This is an analogous mechanism to
the reaction of amino acids with nitrous acid, which gives the alpha-hydroxyacid in dilute conditions and the alpha-haloacid in concentrated acids.
This would explain the products in the papers I linked-- addition to the 4,5-double bond might be preferred for steric or electronic reasons.
I too am interested in what the true oxidizing species in chlorite + HCl mixtures is. You might very well be right that the main reactive species is
chlorine, as evidenced by your chlorocaffeine synthesis.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
3-Hexenoic acid by the reduction of Sorbic acid with Dithionite
6th May 2018
This experiment was inspired by a series of U2Us with the SM member Clearly-not-atara in which we discussed the possibility of sodium dithionite as a
reductant for conjugate double bonds. Sorbic acid was chosen as the substrate for this experiment as it is, for me, the most readily available
compound with conjugate double bonds. At the time the experiment was done I didn't have access to the original work only a schematic overview of
dithionite chemistry from the internet (it looks like part of a slae brochure for a Na dithionite producer!).
The reaction is simple enough:
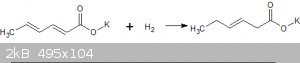
I am not sure about the nature of the counter oxidation product of the sodium dithionite, possibly sodium bisulphite. This would increase the acidity
of the reaction mixture and this may be the reason that the schematic indicated that sodium bicarbonate should be added.
15g of potassium sorbate (0.1 moles) were dissolved in 200ml of water in a 600ml beaker equipped with a magnetic stirrer and heated to 80-90°C.
27.01g sodium dithionite (0.15 moles) and 16g of sodium bicarbonate (0.2 moles) were ground together in a pestle and mortar and then added to the hot
sorbate solution in small portion over about 10 to 15 minutes. After each addition of the solid there was some brief frothing and the solution turned
yellow. Heating and stirring were maintained for about 30 minutes after the completion of the dithionite addition.
The slightly cloudy solution was allowed to cool to room temperature and then acidified with 70ml of 30% hydrochloric acid to about pH 1. The solution
became a cloudy suspension so the mixture was heated almost to boiling but this caused evaporation of much of the intensely “cheesy” smelling acid
so the suspension was cooled to room temperature again. A skin of polymerisation product formed on the surface which was removed and saved for further
investigation (Note 1). The still cloudy suspension was extracted with 50ml of dichloromethane (DCM) but much insoluble material now prevented a clean
separation. The entire volume of liquid was filtered through a 7cm Buchner funnel and placed in a separating funnel (Note 2). Over a couple of hours a
clean separation occurred and the DCM layer was removed, the residue being extracted again with a further 25ml of DCM. The combined DCM extracts were
dried with magnesium sulphate and then the solvent was allowed to evaporate.
When the DCM had evaporated the residue consisted of colourless crystals in an oily acid. The two phases were separated by filtration in a small
Hirsch funnel. The white crystals were very sparingly soluble in water and appear to be unreacted sorbic acid. The oil appears to be the 3-hexenoic
acid, the yield was 5.81g, However, this is probably not a true reflection of the yield as it is volatile and the boiling of the reaction mixture in
contact with air may have caused significant amounts to evaporate or polymerise. The weight of the crystals was not recorded but was <<1g.
Discussion
3-Hexenoic acid exists in two configurations; cis and trans. Both cis and trans isomers are liquids at room temperature (Mps of both are about 12°C)
when pure. However, a mixture of the two is likely to be liquid to lower temperatures. The acid recovered in this experiment remains liquid to about
0-3°. If stored at 4° C for several weeks small colourless crystals form. The small amount a material precluded vacuum distillation with the
equipment that I have. The acid was not investigated further but the strong cheese-like smell is characteristic of this type of acid, both saturated
and mono-unsaturated.
From the experiment above and the observations made subsequently it would appear that slightly more dithionite is required to get complete reduction
and a slightly longer reaction time may also help to improve conversion. The solution should not be boiled after acidification but probably treated
with a filter aid and filtered before extraction with ethyl acetate or ether rather than DCM. The acid is fairly volatile and it is reported that it
can be vacuum distilled without difficult.
Note 1; the skin was dissolved in dilute NaOH solution to give a yellowish solution, charcoal treated and filtered, acidified and extracted first with
DCM and then with ethyl acetate. The later solvent appears to be a better extractant and in spite of the fact it was used after the DCM on evaporation
it left the greater amount of residue. The residue was an acid oil smelling like the main product but a little darker in colour. A little over 1.5ml
resulted. It deposited a few colourless crystals on standing in the fridge for several weeks but neither this nor the main product showed further
tendencies to polymerise.
Note 2; the residue on the filter was found to be mainly sulphur.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Well I have had a go at a Diels-Alder type condensation between recrystallized p-benzoquinone and sorbic acid. I dissolved 0.05 Moles of the former
and slightly more than a molar equivalence of sorbic acid in 65ml of benzene and refluxed for 4 hours. The light amber solution is now cooling but
masses of pale green crystals have formed already. I have based my procedure on the classic Woodward et al paper on the total synthesis of reserpine
(Tetrahedron 1958 p1-65 Yes that's one paper all 65 pages of it!!). He condensed vinylacrylic acid with benzoquinone to obtain two product (isomers),
one a tetrahyronaphthaquinone carboxylic acid and the other an aromatic rearrangement product a dihydro-5,8-dihydroxynaphthalene-1-carboxylic acid.
The former seem to rearrange into the later irreversibly under prolonged heating or under the influence of acids (therefore the former cannot be
Fischer esterified).
Since I am using sorbic acid which has an extra methyl group on the end and I deliberately used prolonged refluxing to force the formation of the
aromatic isomer I hope to have ended up with an extra methyl group in the 4- position relative to the carboxylic acid ie
1,4-dihydro-4-methyl-5,8-dihydroxynaphthalene-1-carboxylic acid. I am not sure how to confirm that this is indeed what I have prepared but I am going
to try to make it fully aromatic by adding bromine across the 2,3 double bond, then de-hydrohalogenating this compound and finally oxidizing it to a
naphthaquinone derivative. More details to follow 
An interesting possibility is the lower temperature condensation of benzoquinone with two moles of sorbic acid to give a polyhydro-anthraquinone
derivative. My next experiment! This need to be done before the initial condensation product aromatises.
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Not particularly helpful, but with reference to the original post, here is what one of the thiobarbituic acid reaction products looks like. This is
actually from an assay of sialic acids and the reactive species is β-formylpyruvic acid not malondialdehyde. The colorant has poor water solubility
and isn't terribly stable and is extracted into cyclohexanone to make measurement consistent. I imagine that oxidation products of sorbic acid produce
a similar color.
https://i.imgur.com/pd18AhW.jpg
FYI: malondialdehyde is a common end-product of free radical oxidation of polyunsaturated fats. The "allyl" position of sorbic acid forms a stable
radical and I imagine follows a similar degradation pattern. https://pubs.acs.org/doi/pdf/10.1021/jf60232a072
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi UC235, I must admit I couldn't see how you would get malondialdehyde anyway. Attach at the double bonds should reduce sorbic acid to C2 or C4 units
so why C3. There again this problem also applies to B-formylpyruvic acid too I guess. The implication is that after the initial attach at either the 2
or 5 position the remaining double bond shifts to the 3 position as it seems to do with the oxidative chlorination.
I have been doing more experiments with sorbic acid and benzoquinone. I have set up an experiment with sorbic acid:benzoquinone 2:1 as a solution in
benzene and I am going to leave it at room temp for a week of two and see if anything happens. I also tried a quick fusion experiment with the same
ratio. I used finely ground sorbic acid and freshly purified benzoquinone and warmed it slowly in a small covered beaker. The mixture started to
darken at the edges then suddenly fused to a brown liquid with the sublimation of a little of the quinone. There was no obvious foaming so the neither
the reactants nor any addition product underwent decarboxylation. Tomorrow I'll try and recrystallise the fused mass and the various products from the
first 1:1 experiment.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I have now worked up the products from my attempts at Diels Alder condensations between p-benzoquinone and sorbic acid. I can confirm that
condensation takes place but the reaction appears slower than that of vinylacrylic acid described by Woodward et al in 1958 (Tetrahedron, 1958,
pp1-57). The products seem to be fairly independent of the ration of benzoquinone to sorbic acid when the reaction was carried out in benzene and in
future I may try using toluene for the refluxed experiments.
I ran three experiments
1- equi-molar amounts of sorbic and and benzoquinone in boiling benzene for 8 hours. Cooled to crystallize and then removed the solvent in stages to
give me 4 fractions and a benzene solution residue.
2- fusion of dry sorbic acid and benzoquinone in the molar ratio 2:1 them extract with sodium bicarbonate solution. This was a mistake as it simply
gave a dark brown solution resembling creosote from which acid precipitates a brown solid which after much careful crystallization gave me a few
colourless prismatic crystals with brown inclusion. The crystals have been preserved but not yet examined further.
3- 2:1 molar ration of sorbic acid:benzoquinone in benzene, sealed in a Schott bottle and kept at 40-45 C for four days and then cooled. The crystals
were collected and then the solvent removed in 2 stages to yield 3 crops of crystals and a small volume of benzene solution as a residue.
Experiments 1 and 3 gave very similar results. The first crop of crystals always consists of mainly white sheaves of Compound A and a little very dark
brown bronzy Compound B. The subsequent crops contained increasing amounts of Compound B with pale yellow diamond shaped crystals of Compound C,
yellow blades of Compound D and in the final crops sometimes a little colourless tabular Compound F. The final benzene solutions were shaken with 1M
sodium bisulphite solution to give a white redution product that is sparingly soluble in both benzene and water Compound E.
Compound A was examined and found after further crystallization from ethanol to melt at about the same temperature as sorbic acid and its solubility
in sodium bicarbonate and it lack of coloured reaction products seem to support the assertion that the compound is unreacted sorbic acid.
Compound B occurs as very dark brown bronzy to black bladed crystals, easily soluble in warm ethanol and bleached to a white sparingly soluble
crystalline material by sodium bisulphite solution (Compound E?). In view of the intense colour I suspect that this is a quinhydrone like compound
perhaps formed from the excess benzoquinone and a C10 quinol derivative resulting from the aromatisation of the D-A condensation product.
Compound C is a bit of a mystery but it appears to be the simple 1:1 condensation product. It crystallizes from both benzene and ethanol in thick pale
yellow diamond shaped tablets.
Compound D occurs only in the later crops and takes the form of deep yellow blades or prisms. After crystallization from ethanol its properties clear
show that it is residual benzoquinone.
Compound E is a reduction product of the residues containing benzoquinone, Compounds B, C and D. It appears to be a mixture of quinol and a less
soluble material, possibly the reduced for of Compound C.
Compound F occurs as colourless plates but is too scarce to permit investigation at present.
Since the yield of Compound C is only about 6% based on benzoquinone I plan to carry out further experiments on a larger scale and using much longer
reaction times and toluene as a solvent to allow higher temperature refluxing.
[Edited on 13-2-2021 by Boffis]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I just tried to scale up the direct combining of sorbic acid and benzoquinone by the solventless fusion process that I tried before but at 10x the
scale. With less than one gram of reactants in the original experiment the reaction was quiet with no obvious reaction apart from the melt becoming
dark. I ruined this experiment by trying to leach the reaction mixture with sodium bicarbonate and obtained a dark brown creosote like liquid.
I tried this again with 5g of each reactant, roughly 1:1 molar ratio, ground together first and then gently warmed over a free ethanol flame in a
small round-bottom flask. The mixture melted quite suddenly to a rich orange liquid. Then while I was holding the flask but not in then flame any
more, the mixture began to darken and then suddenly erupted emitting copious amounts of acrid purple smoke which condensed to a purple dust on
everything around about. No actual damage was done but I wasn't 'alf surprised
and I have a bit of a clean up!.
So beware, if you decide to try this reaction without solvent!
The toluene solvated reaction is currently refluxing away quietly.
Update:- well I refluxed the toluene solution for 9 hours and then let it cool. This morning I found some yellow-brown-orange crystals in a light
brown liquid with a rim of soft gloopy tar at the top of the liquid level. I warmed the flask and poured the contents into a beaker, this left behind
most of the gloopy tar, another gift from chemistry hell, in the flask. The flask is now full of chromate sulphuric acid mixture and the contents of
the beaker crystallising ontop of a layer of yet more tar.
I think the conclusion of yesterdays experiments is that the secret is patience and reaction temperatures at or below 80 C.
I have also recrystallised all of the compound C and obtained a small quantity of very pale yellow aggregates of diamond shaped crystals, currently
drying.
[Edited on 17-2-2021 by Boffis]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I have a jar or 1,2,3,6-tetrahydrophthalic acid:

Does anyone know if this compound is a dienophile like maleic anhydride? Would it be better as an ester rather than a free acid.
My plan is to try and condense it with sorbic acid to form a hydro-8-methyl-naphthalene-2,3,5-tricarboxylic acid. Possible?
|
|
|
Pumukli
National Hazard
Posts: 705
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
I'm not sure but I think I saw it in a "sorbic acid paper" that it can be condensed with sorbic acid (or its Me-ester) to form that compound...
(Years ago I was contemplating this synthesis and DO remember drawing structural sketches of the compound on a piece of paper.)
Tetrahydro phthalic acid is a precursor of captan and it was probably extensively investigated in the '50-ies.
|
|
|









{kind=link}