Lion850
National Hazard

Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
Report on making Tin Iodide and Antimony Iodide
After seeing videos on YouTube I wanted to make Tin Iodide SnI4. It turned out to be reasonably straightforward. I made 2 batches.
First:
- 200ml of dichloromethane
- 8g Iodine
- 6g Tin granules
Contents placed in a 500ml beaker. A 500ml RBF filled with water was placed on top of the beaker (RBF supported by a stand) to act as a simple reflux.
The DCM was brought to a gentle boil with magnetic stirring. After 45 minutes not much happened, I then lifted the RBF to let the DCM escape and thus
reduce the overall volume. Once the volume was under 100ml the color started clearing, by the time it was down to around 70ml it was a nice orange
with no more sign of Iodine. I filtered it off into a small beaker and boiled it down to 20ml, at which point there was already a substantial amount
of orange precipitate. I cooled the beaker in an ice bath, and filtered again. I then dried the red SnI4 crystals with paper towel and by simply
leaving it out in the (hot) shed. The first run gave 8g of product.
Second:
There was still a lot of unreacted Tin granules so I added another 8g Iodine, and this time only 100ml DCM. The solution was allowed to boil down
slowly and by the time it reached approximately 60ml, about 40 minutes later, the reaction was complete and the SnI4 was concentrated and recovered as
above. This time the yield was 8.6g, but I am not 100% sure it was completely dry, may be a gram or so of DCM.
On YouTube I saw PoorMans Chemist' video where he made beautiful red Antimony Iodide using Toluene as solvent. I decided to try using DCM:
- 8g Iodine
- 5g Antimony powder
- 100ml DCM
After 2 hours of boiling nothing appeared to have happened. Solution stayed dark. PoorMans Chemist in fact told me that DCM may not work as Antimony
needed a higher temperature, and that if it did not work to try Toluene or Xylene. So I then chucked out the reagents. But, I did see some traces of
red when pouring it out....
I then went to the local hardware and got Xylene (Toluene was not immediately available near me). I started again with 7g Iodine, 4.5g Antimony
powder, and 100ml Xylene. I boiled this for 2 hours (bp should be around 110 deg as per Xylene spec) but the solution stayed pitch black. I then again
removed the "refluxing" RBF and allowed it to boil vigorously (always with stirring) down to around 60ml. Still pitch black. I was just about to give
up and chuck the contents when I noticed 2 things: There was red spots on the sides of the beaker and then fast stirring splashed the contents up the
liquid was only black with no more purple like earlier. I then wondered whether the very fine Antimony powder which was purposely in excess was
staying in suspension and making the solution look black? I did a filtration of the boiling liquid (by now reduced to under 50ml) and was rewarded
with a blood red filtrate with red crystals of SbI3 settling as it started to cool! I cooled it in an ice bath, filtered off the crystals, and dried
on the filter paper on a slightly hot hotplate. The result was 4.6g of very beautiful red crystals with what appears to be golden specks. In fact it
reminded me of the lead iodide golden rain. The photo does not really do it justice.
Next to try to make lead iodide and manganese iodide this way. I'm going to try to attached some photos, hope it works.
   
|
|
|
Lion850
National Hazard
Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
Hi guys when I view my photos in my above post on my laptop they are lying on their sides....how to turn them the correct way?
Thanks, Leo.
|
|
|
j_sum1
Administrator
Posts: 6368
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
This is pretty cool, Lion850. It does not sound like an overly challenging synthesis but a dramatic result.
Thanks for sharing. (And don't worry about the pictures.)
|
|
|
Texium
|
Thread Moved
24-11-2019 at 06:22 |
AJKOER
Radically Dubious
Posts: 3026
Registered: 7-5-2011
Member Is Offline
Mood: No Mood
|
|
Nice experiment!
-----------------------------
FYI, comments from atomistry.com on SnI2 (at http://tin.atomistry.com/stannous_iodide.html which is constructed from extracts from old chemistry journals):
"Stannous Iodide, SnI2, is obtained by adding potassium iodide in slight excess to a concentrated solution of stannous chloride, or by the action of
hydriodic acid on tin; it crystallises in orange-red octahedra, which may be obtained by melting the compound, or evaporating its solution in carbon
disulphide; but it is probably dimorphous. It melts at 320° C., and boils at 720° C. The dihydrate SnI2.2H2O is said to exist; 100 parts of water
dissolve 0.93 parts of anhydrous stannous iodide at 20° C., and 4.03 parts at 100° C. It is much more soluble in hydriodic acid and alkali halide
solutions, owing to the formation of a complex acid or salts."
[Edited on 3-12-2019 by AJKOER]
|
|
|
woelen
Super Administrator
Posts: 8071
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Very nice synthesis of SbI3 and SnI4. I made SnI4 a few years ago:
https://woelen.homescience.net/science/chem/exps/SnI4/index....
Recently I made a somewhat larger batch of SnI4 and made a display sample of the material in a sealed glass ampoule.
----------------------------------------------------------------------------------------------
I also did an experiment with SnI2. The latter can be made in aqueous solution, although I think that isolation of the pure compound will be much
harder than the isolation of SnI4.
https://woelen.homescience.net/science/chem/exps/tin+iodide/...
|
|
|
Lion850
National Hazard
Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
Hi Woelen I will have a go at SnI2 when I have the time.
|
|
|
Lion850
National Hazard
Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
Some 9 months since I made the Tin (iv) iodide SnI4, and after watching PoorMans Chemist's recent video where he makes tin (ii) iodide SnI2 by double
displacement in water I thought it would be nice to have both iodides so also had a go. The link to PoorMans Chemist's video:
https://www.youtube.com/watch?v=fR1pem_ytYE&t=846s
I thought this would be as straightforward as they come but there were a few surprises. Expected double displacement reaction:
SnCl2.2H2O + 2KI = SnI2 + 2KCl + 2H2O.
- 7g tin chloride added to a small beaker with 30ml water & stir
- Solution stays cloudy, but if filtered the filtrate is clear and there is a white remainder.
- Add 10 drops concentrated HCl, still cloudy.
At this point I stopped to read up more...and found out that SnCl2 must always be dissolved in a small amount of water - in excess water it hydrolyses
into an insoluble salt. Duh....start again.
- 7g tin chloride in 5.5g water and stir....almost clear solution!
- Dissolve 13.5g KI in 12g water, clear solution.
- Add the KI solution to the SnCl2 solution while stirring
- Yellow thick emulsion, stir bar stuck! Some orange spots forming.
- Add water slowly while stirring with a glass rod until the solution thinned enough to be moved with the stir bar.
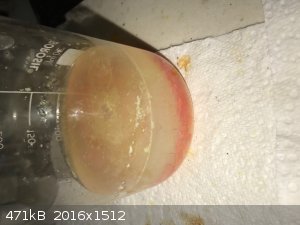
- Mainly orange with some yellow lumps. Heat to about 50C while stirring, now mainly orange with some yellow on the edge and smooth. See photo below.

- Gravity filter. Orange remainder but more yellow on top, pale yellow filtrate.
- Wash the remainder with water in the funnel and transfer to steam bath to dry. Photo below.

- Color darkens a bit when hot (dry) but more orange when cool. 4g dry mainly orange powder, which was a poor yield.
Usually I clear up before leaving the shed for the night but I was called into the house and left the cleaning for the next day. Which was lucky,
because the next day I saw that red crystals settled out of the beaker that has the yellow remainder. See photo below.
Some of the SnI2 crystals were obviously fine enough to pass through my filter paper. I decanted the supernatant liquid, rinsed the crystals with
water and decanted again, and then added this crop to the crystals drying on the steam bath. This second crop added a gram or so to the eventual dry
weight, and became more orange as they dried. Photo below of the wet crystals that crystalized out overnight.

After drying on the steam bath, the color of the cool dry product was a lighter orange. The photo below shows the final color on the left. The right
bottle is the SnI4 made 9 months ago and stored in this clear bottle; it has held up quite well but has darkened a bit.

When PoorManChemist did his reaction he also briefly got a yellow solution. I think the SnI2 appears yellow when in dilute suspension or when the
solid is very fine. Once it clumps together it is more orange, and if wet red - but this is just my guess.
If I were to do this again I would make the KI solution a bit more diliute and see what happens if the SnCl2 is added to the KI instead of the other
way around. My reaction probably had too little water once mixed and thus I initially ended up with the thick yellow emulsion. Again, I am just
guessing.
The purity and appearance of the final product could probably be improved if it was dissolved and recrystallized, and then dried slowly at lower
temperature instead of on the steam bath.
|
|
|
Lion850
National Hazard
Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
I needed to make more antimony iodide and thought I would post the process again as it seemed much easier this time around. And it is such a beautiful
salt!
- 200ml Xylene in beaker
- Add 7.5g fine antimony powder
- Add 15g iodine crystals
- Watch glass on top of beaker filled with water - to reduce loss of xylene while boiling and watch the color of the condensed drops
- Bring to very gentle boil on hot plate while stirring. For a short time iodine fumes were visible in the beaker, and the drops condensing on the
watch glass were purple. Photo below.

- After 30 minutes the drops were clear, and no more sign of purple in solution or solution splashes.
- Continue to boil for another 30 minutes. Loss of xylene was very little; some smell of xylene in the shed.
- Stop boil and immediately gravity filter while near boiling. Red solution, and SbI3 crystalizing out on cold surfaces. Unreacted antimony powder in
beaker. Photo below.

- Let solution cool to room temperature. Decant supernatant liquid to a separate "2nd crop" beaker. Scratch out crystals, red with gold specs. Photo
below.

- Dry between sheets of filter paper and paper towel, then dry in hot sun (under a steel dish) for 2 hours.
- 9.9g of dry crystals from the first crop. Photo below.

- I then tried to recover all the crystals that was left behind in the funnel and in the filter and boiled them again with the supernatant xylene
solution previously recovered. This was to be the second crop. I boiled the solution down from 150ml to under 50ml on the hotplate and then steamed it
dry on a steam bath, just to see what happens. I got a different looking crop of crystals, not nearly as beautiful. Photo below.

- I then boiled the 2nd crop crystals with 80ml water for 30 minutes. This gave a yellow ppt which according to Atomistry is SbOI, antimony oxyiodide.

- The solution was gravity filtered and the yellow ppt washed in the filter with boiling water.
- The product was well stuck / soaked into the filter paper, it was difficult to remove! Only a gram or so was recovered. Photo below.

Cleaning: This stuff stains porcelain. I tried cleaning the evaporating dishes with acetone, xylene, and concentrated HCl but they were still
coloured. I then found that a NaOH solution removes the stain very easily.
These SbI3 crystals keeps very well. The smaller batch I made nearly a year ago and which was kept in a clear bottle still looks very much the same as
when made.
|
|
|
MidLifeChemist
Hazard to Others
Posts: 192
Registered: 4-7-2019
Location: West Coast USA
Member Is Offline
Mood: precipitatory
|
|
Thanks for reposting. I may try this one day. I have Iodine and I have Antimony shot arriving in a couple of days. I think I can buy Xylene in the
hardware store.
But I don't have Antimony powder. Has anyone converted Antimony shot (pellets) to powder before? Not sure if I should try it chemically, or by some
physical process like sanding or grinding somehow.
|
|
|
teodor
National Hazard
Posts: 984
Registered: 28-6-2019
Location: Netherlands
Member Is Offline
|
|
@MidLifeChemist As a side note, be careful with antimony. On strong reduction (for the sake of safety, let's say in all the cases when you reduce a
compound to the element) you can get SbH3 which is supposed to be a blood toxin with extremely low amount, according to a theoretical toxicological
data, required to make you ill or dead.
[Edited on 4-12-2020 by teodor]
|
|
|
Lion850
National Hazard
Posts: 517
Registered: 7-10-2019
Location: Australia
Member Is Offline
Mood: Great
|
|
Midlifechemist just go with the shot. It will probably just take quite a bit longer, maybe better then to do under proper reflux setup so it can run
with the boiling xylene for hours if necessary.
|
|
|









