| Pages:
1
2
3 |
xdragon
Harmless

Posts: 9
Registered: 4-8-2020
Member Is Offline
|
|
I don't have the exact protocol anymore, but benzoquinone can be fairly easily synthesised by the oxidation of hydroquinone dissolved in IPA by 1 eq.
Oxone in aq. solution, catalysed by 10 mol % NaBr and if needed a bit of heating. KBr will likely not work. This may be interesting for people who do
not have access to iodine or useful H2O2 concentrations. Completion of reaction is fairly easily judged by disappearance of the ugly dark colour from
the quinhydrone complex.
Some literature on which those findings were based:
(1) Yakura, T.; Ozono, A.; Morimoto, K. An Efficient Catalytic Oxidation of P-Alkoxypenols to p-Quinones Using Tetrabutylammonium Bromide and Oxone.
Chem. Pharm. Bull. 2011, 59 (1), 132–134. https://doi.org/10.1248/cpb.59.132.
Also, thanks for your quality write-ups and pictures, Benignium. I'm getting excited for the 4-Et-2,5-DMBA, I got some of the acetophenon but haven't
had the time to deal with it yet.
[Edited on 8-11-2022 by xdragon]
[Edited on 8-11-2022 by xdragon]
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
You're most welcome, xdragon! 
Great contribution! I've also wondered about the utility of persulfates in this regard, but unfortunately, I haven't looked into it yet.
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
I'd like to formally welcome everyone to what could become the longest page on Sciencemadness! Not particularly desirable, but then again, neither are
the world's longest fingernails, and yet someone has to have them.
2,5-dimethoxy-4-ethylbenzaldehyde
194.23 g/mol

This process of forming 2,5-dimethoxy-4-ethylbenzaldehyde,[1] while reasonably established and straightforward, involves some elements that
I found quite intimidating to plan for: in between the more familiar steps of methylation and formylation, there were two classical manipulations
that, aside from being efficient in forming the desired alkyl substituent, coincidentally displayed tendencies to demethylate the neighboring methoxyl
on the side. Fortunately, ventures outside PiHKAL to browse the usual discussion boards revealed that improved results could be obtained via simple
modifications to make the reaction conditions milder.
Moreover, reports of unpredictability and poor yields, from those who had performed the final conversion to an aldehyde, had left me anticipating more
of an eventual impediment. However, I was delighted to find out that my impression was substantially inaccurate.
[1]: PiHKAL: #24 2C-E. https://isomerdesign.com/PiHKAL/read.php?domain=pk&id=24
[b1] 1,4-dimethoxybenzene
138.16 g/mol
The methylation of 4-methoxyphenol seems to be about as pleasant as methylations get: the phenol is readily converted, and not terribly fussy when it
comes to oxidation, allowing for good yields of a remarkably fragrant[1] product, and doubtlessly the most satisfying substance that I've
steam distilled to date.
[1]: The odor is quite unique, but there's something very familiar about it. I suspect that much of this familiarity stems from its
resemblance to some species of clover that was (or were) abundantly present in my childhood.
Experiment 1
In a 250 mL round-bottomed flask, 4-methoxyphenol (20.16 g, 162 mmol) was dissolved in acetone (46 mL). Potassium carbonate (27.50 g, 199 mmol) was
added to the resultant solution, and the mixture was stirred for 50 minutes prior to adding dimethyl sulfate (20.2 mL, 213 mmol). The mixture was then
heated to reflux and shortly allowed to cool back down.[1] Stirring was continued for 20 hours,[2] after which there was a
four-hour period of further refluxing. The mixture was allowed to stand for a further 42 hours[2] before the caked solids were broken up by
adding, roughly 30 minutes apart, two 10 mL portions of water and an aqueous 10% solution of sodium hydroxide (4.1 g, 103 mmol).
[1]: My intention was to keep refluxing the mixture, but my mantle didn't register the low power setting that I tried to use, and
heating was thus discontinued by accident.
[2]: At ambient outside temperatures, fluctuating on either side of 20°C.
[Fig. 1] Solution of MeHQ in acetone being stirred over K2CO3

[Fig. 2] Above mixture after 26 minutes of stirring

[Fig. 3] Reaction mixture following addition of DMS

[Fig. 4] Above mixture after 23 hours, during fourth and final hour of reflux

[Fig. 5] Caked reaction mixture following addition of 10 mL of water

Work-up (1)
After the solid mass in the reaction vessel had been broken up, and the suspension had been stirred for a while, the mixture was vacuum filtered. The
filtered solids were washed with a few small portions of acetone. Acetone was then removed from the filtrate by boiling/evaporation under vacuum —
initially without heating, until the temperature of the filtrate had reached ~0°C, and then on a 100°C hotplate until it reached 10°C. The mixture
was vacuum filtered and the filter cake was dried overnight in open air to yield 19.40 grams (86.5%) of the crude product as brown platelets.
The brown material was mostly dissolved in 25 g of 36–40°C heptanes, leaving 0.53 grams of an insoluble tar in the flask. The flask was rinsed with
5 grams of fresh solvent that was gravity filtered through cotton wool along with the previous portion. The mixture was allowed to stand in a beaker,
under a perforated piece of cling film, until it had lost ~11 grams of its mass. This was then cooled to somewhere below 0°C in the freezer, and
vacuum filtered to obtain 17.45 g (77.8%) of cream-colored crystals.
Finally, steam distillation of the product from 350 mL of water was attempted. Initially, the short path still head that was used had cool water
running through the condenser, which rapidly caused it to become clogged. Using 46°C condenser water (which gradually warmed up to 54°C on its own)
allowed the steam distilled product to remain fluid until it was deposited, but necessitated the use of a jointed collection flask as some vaporized
product also came over. Once the impure mixture had been depleted of product, the collected distillate was warmed to melt the solidified product, and
pitched into a beaker. Filtration of the solidified product gave, after 18 hours of air-drying, 14.25 g (64.1%)[1] of 1,4-dimethoxybenzene
as colorless, crystalline chunks.
[1]: A significant portion of the obtained product was lost to evaporation.
[Fig. 6] Post-reaction mixture after breaking up of caked solids

[Fig. 7] Solid product precipitating from vacuum filtrate as acetone boils off

[Fig. 8] Impure crystals of 1,4-dimethoxybenzene

[Fig. 9] 1,4-DMB crystallizing from heptanes <> Heptane-insoluble tar

[Fig. 10] Steam distillation of impure product

[Fig. 11] Premature freezing of steam distilled 1,4-DMB

[Fig. 12] Deposition of gaseous product in receiving flask

[Fig. 13] Purified 1,4-dimethoxybenzene

Experiment 2
In a 1000 mL two-necked RBF, there was placed water (395 mL) and NaOH (18.40 g, 460 mmol). To the stirred, degassed[1] alkaline solution, a
separately prepared solution of mequinol (38.15 g, 307 mmol) in acetone (50 mL) was added. Lots of white solid precipitated, and remained undissolved
until the addition of a further 50 mL of acetone (somewhat surprisingly) redissolved most of it. Me2SO4 (38 mL, 400 mmol) was
then added from a separating funnel at a rapid dropwise pace; the mixture became opaque with precipitate, and the stopcock was fully opened to allow a
more exothermic reaction to prevent any solid from negatively affecting the weak magnetic stirring of the heating mantle. A less dense, immiscible
liquid phase formed, and the mixture was stirred for a further 4 hours until the formed product had crystallized.
[1]: Degassing was performed by pulling an aspirator vacuum over the stirred mixture twice, each time maintaining it for 5 minutes
before normalizing the pressure using argon.
[Fig. 14] Appearance of poorly soluble 4-methoxyphenolate

[Fig. 15] Dissolution of phenolate due to additional acetone

[Fig. 16] Reaction mixture following addition of DMS

[Fig. 17] Crystalline product in alkaline post-reaction mixture

Work-up (2)
NaHCO3 (5 g, 60 mmol) was added to neutralize excess hydroxide, and a direct steam distillation of product from the reaction mixture was
attempted. 60 mL of the initial distillate was collected (until two immiscible liquid phases were observed in the condensing vapor) and moved
aside.[1] Once distillate was being collected at 90°C, the condenser water was heated up to 50°C, and the distillation was continued
until (after collecting ~400 mL) no more product was observed in the distillate.
The steam distillate (collected directly into a separatory funnel) was extracted with dichloromethane — using just enough to dissolve all of the
solid product, followed by a second portion of just a few milliliters. Most of the solvent was then atmospherically distilled off in a warm water bath
prior to continuing its removal for a moment under reduced pressure. On cooling, the mixture crystallized partially, and it was poured into a
recrystallization dish where it was stirred around by hand, under a steady flow of air, until there was obtained a crunchy solid that seemed perfectly
dry. The dish was covered with a sheet of paper towel and kept as such for a few hours to afford 38.08 grams (89.7%) of a colorless, crystalline
product melting at 54.3–56.6°C (lit. 54–56°C).
[1]: I believe that I discarded this head portion, but there are no records of its fate.
[Fig. 18] 1,4-dimethoxybenzene crystallizing from saturated DCM

[c1] 2,5-dimethoxyacetophenone
180.20 g/mol
Next, the 1,4-dimethoxybenzene is subjected to a classic Friedel-Crafts acylation — acetyl chloride is treated with aluminium chloride to form a
strongly electrophilic acylium carbocation that gets attacked by the aromatic ring, yielding an arenium carbocation that then gives up a proton, thus
re-establishing aromaticity, generating hydrogen chloride, and regenerating aluminium chloride.
The presence of HCl and AlCl3 makes for a harsh environment that is capable of methoxyl cleavage, giving rise to a
2-hydroxy-5-methoxyacetophenone side product. The reported yield of 2,5-dimethoxyacetophenone in PiHKAL is 77.8%, which is far from terrible, but
seems possible to improve upon by taking more time to perform the reaction at lower, more controlled temperatures; a report by the Hyperlab user
Pine_tar[1] seemed to suggest that a dropwise addition during active cooling in an ice bath, followed by maintaining the reaction mixture
at 4°C for a further six hours, would produce next to no phenol. My intention was to find out, but my quest for evidence had its fair share of flaws.
My first experiment appeared to have been quite successful, up until a rather farcical vacuum distillation which compromised the yield as well as its
purity. Luckily, I still had a bunch of mequinol, whose methylation to provide for a second experiment was no issue.
As an unwelcome surprise, I found my sample of aluminium chloride to be quite contaminated, from having completely eroded the material that was
serving as the cap liner of the storage (and retail) bottle. With no real options to remedy the situation at hand, I ended up using the (crudely
mechanically cleaned) contaminated material. Fortunately, it turned out to be viable, and I eventually decided to use it for the second experiment as
well.
[1]: https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
Experiment 1
In a 250 mL round-bottomed flask, in an ice bath, acetyl chloride (10 g, 127 mmol) was pipetted as a slow stream to a stirred suspension of aluminium
chloride (17.63 g, 131 mmol) in DCM (50 mL). A separately prepared solution of 1,4-dimethoxybenzene (13.71 g, 99 mmol) in DCM (33.5 mL) was then
pipetted in dropwise over ~15 minutes.[1] The mixture was stirred for a further 126 minutes, until all of the ice in the bath had
melted,[2] before being stored for 4 hours in the refrigerator, followed by 20 hours in the freezer, below -20°C.
[1]: Both additions were quite exothermic, producing plooms of vapor as solvent boiled off. For the addition of the benzene in
particular, the use of an addition funnel would have been ideal.
[2]: The temperature of the reaction mixture at the time of its removal from the bath was ~6°C.
[Fig. 19] Aluminium chloride of questionable integrity

[Fig. 20] Above aluminium chloride under dichloromethane

[Fig. 21] Reaction mixture following addition of acetyl chloride

[Fig. 22] Reaction mixture after adding ~20% of all 1,4-DMB

[Fig. 23] Reaction mixture after adding all 1,4-DMB

Work-up (1)
The chilled reaction mixture was quenched by pouring it into a 250 mL separatory funnel containing 125 mL of cool water; there was an exothermic
reaction, and some DCM boiled off. Once the exotherm had subsided, the biphasic mixture was shaken, and the organic layer was harvested. Two more
extractions were performed using 10–15 mL portions of DCM, and the combined DCM partitions were washed with three 20 mL volumes of 5% aqueous NaOH.
The organic extract was concentrated by distillation from a warm water bath to afford 28.63 grams of a clear, brown liquid that was distilled under
aspirator vacuum. An initial 1.49 g of a low-boiling, low-viscosity fraction with a uniquely sweet and hydrocarbonesque smell was collected (and
allowed to evaporate). Following the largely unsuccessful vacuum distillation and a subsequent remedial steam distillation,[1] there was
obtained, by extraction using DCM, 14.93 g (≤83.5%) of a discolored liquid with a faint, agreeable aroma.[2]
Acidification and DCM extraction of the pooled alkaline washes gave, on evaporation, ~70 milligrams of an oily, yellow residue with a pleasant odor
which resembled a mixture of carrot and root beer. This was discarded.
[1]: The vacuum line was leaking, which led to some visible scorching of the crude product. The leak was located and wrestled shut by
hand, which caused an improperly tightened connection on the water pump to loosen and eventually become disconnected near the end of the distillation,
spraying water in my face and allowing a backflow of water into the distillation apparatus. Some of the collected distillate was sucked back into the
distillation flask with water, and some (<20%) was ejected through the vacuum line as that water then flash boiled. Fortunately, the water was
pure, and none of the glassware broke. The dark red oil in the distillation flask was steam distilled to reclaim some of the sucked back product.
[2]: Bearing resemblance to unsubstituted acetophenone as well as 2,5-dimethoxytoluene, with a definite presence of some unreacted
1,4-dimethoxybenzene.
[Fig. 24] Reaction mixture, fresh out of the freezer

[Fig. 25] Pooled DCM phases

[Fig. 26] L to R: Alkaline washes, DCM-extracted aq. mixture, DCM extract

[Fig. 27] Concentrated DCM extract prior to vacuum distillation

[Fig. 28] Aftermath of blundered vacuum distillation

Experiment 2
A 500 mL two-necked round boiling flask was set up in an ice bath,[1] and a powerfully stirred suspension of AlCl3 (38.69 g, 290
mmol) in DCM (109.6 mL) was materialized inside it. Using a pipette, AcCl (16.8 mL, 236 mmol) was added dropwise over 10 minutes, and the mixture was
then allowed to stir for a further two minutes or so before commencing the dropwise addition of a solution of 1,4-DMB (30.06 g, 218 mmol) in DCM (71.5
mL) from a 250 mL addition funnel. After 20 minutes, the addition was paused; the reaction mixture was allowed to cool for 5 minutes, and an
additional portion of AcCl (3.1 mL, 44 mmol) was pipetted into the mixture.[2] The addition was then resumed for 75 minutes before pausing
again to re-establish the magnetic stirring that had failed up to 10 minutes prior.[3] After about an hour had passed, with the stirring
enabled once more, the remaining ~3 mL of solution was added from the funnel over a period of 9 minutes. Two hours after completing the addition, the
cooling bath temperature was measured to be 6.5°C, and more ice was added. Stirring was then continued for 40 minutes.
[1]: Water was recirculated down the sides of the flask from below crushed ice using a submersible 12V water pump.
[2]: This portion of acetyl chloride was to be included in the initial addition, but there was a miscalculation. Whatever impact this
late addition had doesn't seem to have been terribly detracting.
[3]: A DIY stirrer (elaborated on below) was used to agitate the mixture. Its distance from the ice bath was minimized by using an
opportunely proportioned Donald Duck pocketbook as a spacer underneath it. An unforeseen consequence of this was the swelling that took place as the
comic absorbed droplets of condensation falling from the underside of the bath, eventually jamming the stirrer. This meant that, for ≤10 overlooked
minutes, the benzene was added to a stationary mixture. Not great, but as it turned out, not terrible, either.
Work-up (2)
The reaction mixture was poured in 300 grams of crushed ice, and the resulting biphasic mixture was stirred for 15 minutes. The dense organic phase
was collected, and the aqueous phase was extracted twice more with 30 mL portions of DCM. After combination, basic washing with aqueous 5% NaOH (3x40
mL), and distillation of the extracts to remove the solvent, a residual amber liquid was obtained from the organic extract. This was then distilled
under reduced pressure at ~170°C to yield 30.45 g (77.8%) of a yellow oil with a measured density of ~1.14 g/cm3 at 20°C,[1]
and a familiar, faint aroma which implied a total absence of the starting material. This product was used in the subsequent synthetic
step.[2] 5.13 g of a dark brown oil was retained from the distillation flask, but not processed further.
Acidification and organic extraction of the pooled alkaline washes gave a little over 1.5 grams of a yellow solid that had the previously encountered
odor of root beer and carrots, a tendency to fuse with polystyrene, and a wide melting point range of 37–45°C; this was the crude
2-hydroxy-5-methoxyacetophenone side product.[3]
[1]: Fisher Scientific: Literature value of 1.1300 g/cm3. https://www.fishersci.com/shop/products/2-5-dimethoxyacetoph...
[2]: The subsequent reduction took place (starting) 49 hours after this vacuum distillate was photographed (fig. 35) and stored in a
100 mL amber glass bottle. Strangely, when I proceeded to pipette it into the reduction mixture, I saw that it had become quite dark and brown in
color. The remaining portion darkened further as time progressed, and was combined with the visually unchanged product from the first experiment
pending a second distillation.
[3]: The sample whose melting point I report had been stored in a polystyrene cup for a month, crudely scraped off of the maimed
plastic, stored in a cling-film-covered 25 mL beaker for two months, filtered as a solution in minimal MeOH, and obtained as a waxy residue on a watch
glass (fig. 34) following the evaporation of said MeOH.
[Fig. 29] Reaction mixture next to crushed ice

[Fig. 30] Reaction mixture being poured in crushed ice

[Fig. 31] Quenching mixture being stirred

[Fig. 32] Merged organic partitions <> Aqueous partition

[Fig. 33] Combined alkaline washes being acidified

[Fig. 34] Crude phenolic side product

[Fig. 35] Freshly vacuum distilled 2,5-dimethoxyacetophenone

[d1] 2,5-dimethoxy-1-ethylbenzene
166.22 g/mol
Reduction of the acetyl moiety to obtain the desired ethyl substituent is commonly performed in one of two ways: via the Clemmensen reduction, which
employs amalgamated zinc and hydrochloric acid; or by using the Wolff-Kishner reduction where, in strongly basic conditions, a deprotonated hydrazine
condenses with the carbonyl carbon, eliminating water to form a hydrazone intermediate whose diimide tautomer collapses on deprotonation, liberating
N2 and affording a carbanion that is then rapidly protonated. Both methods share the tendency to cleave methoxyls. Although I chose the
latter, I would at some point like to attempt using the Clemmensen on the surplus of produced acetophenone, to find out if the mercury can be omitted
for results that are comparable, or perhaps replaced by another alloy, such as one that contains zinc and gallium.
In PiHKAL, the described procedure takes place in triethylene glycol, and the reaction mixture is refluxed at 210°C for 3 hours. This has been
criticized as too harsh for 2,5-dimethoxyacetophenone, and cited as the primary reason for the meager reported yield of ≤22.0 g (≤23.9%) of
2,5-dimethoxyethylbenzene from 100 g (555 mmol) of the starting material, with an additional 28 g (30.4%) recovered via isolation and remethylation of
the 2-ethyl-4-methoxyphenol side product. As I had no reason to dispute these claims, I decided to plan and execute accordingly.
For my experiment, I opted to go with monoethylene glycol, as that was what I had on hand, having neglected to acquire beforehand the diethylene
glycol which others seemed to have preferred. I surmised that with its boiling point of 197°C, the monomer would prove advantageous in helping to
ensure that the mixture doesn't get too hot when it boils, and, after seeing the results, I believe that I was largely correct. It turned out that my
issues lay elsewhere.
The very first issue encountered was the poor solubility of alkali in MEG. NaOH was chosen over the similarly soluble KOH due to its lower molecular
weight and higher apparent purity,[1] and a a small quantity of water, extrapolated from documented instances of the use of hydrazine
hydrate, was incorporated with the goal of achieving a consistency that wouldn't overwhelm the modest torque and magnetic power of my heating
mantle.[2]
The second issue has to do with a problematic foaming, which, by some divine intervention, persisted on the cusp of ruining things, yet never
escalated. The foam appeared to consist of bubbles that were, for the most part, remarkably small, and as someone who enjoys Guinness enough to
occasionally purchase it in cans,[3] my immediate thought was that perhaps I was witnessing the evolution of nitrogen — and this would
seem to make some sense timing-wise. As to the extent to which nitrogen causes or exacerbates the foaming, I can only guess; it's also possible that
the foaming was more or less specific to the use of MEG, or that my particular MEG, obtained via the fractional distillation of an engine coolant,
contained culpable contaminants (this could also explain the pronounced yellowing on dissolution of NaOH, which struck me as odd). In any case, I'd
like to learn how to prevent the foaming, since it clearly prolongs the contact of glassware with the highly caustic (wet) reaction mixture.
[1]: My new (i.e. old, but unopened) sample of KOH turned out to be quite green for some unknown reason. The moisture content of KOH
also tends to be higher.
[2]: Normally, I would have resorted to using my trusty Corning hotplate, but, unfortunately, we developed a prohibitive
fault during the summer. The hotplate remains in disrepair to this day, but the void in its place has inspired some nifty solutions, namely the
incorporation of PC-fan-based DIY magnetic stirrers in conjunction with accurately adjustable hot water baths utilizing a sous vide immersion
circulator.
[3]: Guinness is a beer that is known for its fine, creamy effervescence. To achieve this, the cans are pressurized by the inclusion
of liquid nitrogen inside a "floating widget" — a cryptic sphere which inspired my curiosity. Shout-out to Google.
Experiment 1
Monoethylene glycol (126 mL) was situated in a 250 mL two-necked round boiling flask, and to it, in an ice bath and with magnetic stirring, NaOH
(29.95 g, 749 mmol) was added in portions, along with some water (7.35 g in total) to aid its dissolution. A sufficiently stirrable mixture was
pursued for nearly five hours: not everything dissolved, and the subsequent addition of hydrazine sulfate (32.86 g, 253 mmol) had no perceivable
effect on the high difficulty of agitating the mixture; a desireable thinning was achieved by immersing the flask in a 60°C water bath, and once the
bath temperature had been brought up to 80°C, 2,5-dimethoxyacetophenone (14.64 g, 81 mmol) was added via pipette. The flask, fitted with a Liebig
condenser, was then moved into a 1000 mL heating mantle where it was immediately heated toward reflux with magnetic stirring. After refluxing the
mixture for 30 minutes, the heating was discontinued for two hours[1] prior to replacement of the Liebig with a water-cooled short path
still head and the initiation of a delicate distillation to remove water: on boiling, a ~120°C foam appeared, and promptly climbed into the
fractionating portion of the still head, despite attempts to suppress it by adding more MEG (20 mL was added in total) from a separatory funnel, as
well as adjusting insulation and the application of heat.[2] Fortunately, the foaming readily equilibriated such that it never reached the
condenser, and after observing no erratic behavior for three hours, the distillation was left to run unattended for two consecutive three-hour
periods.[3]
After the six-hour period, the mixture was no longer foaming or boiling; the collection flask was emptied,[4] and heating was increased to
raise the temperature of the mixture to 140–150°C, resuming the collection of distillate. After an hour, with the mixture now at ~170°C, only
intermittent condensation of 140°C vapor took place in the still head, and the condenser was emptied of most water before incrementing the heat.
Soon, the collection of distillate picked up again (to a rate of approximately one drop every two seconds), and a coolant tube was repurposed to
observe gas exiting the system at a leisurely pace[5] as the collection temperature climbed past 180°C, eventually reaching 197°C before
the reoccurrence of foaming made the short-lived distillation of glycol unsustainable. Combating the foam by adding fresh MEG (10 mL) proved futile
once again, and the heating was stepped down, maintaining the mixture at ~190°C until 90 minutes had passed since distillate was first collected at
>180°C. At last, the heating was discontinued, and, after concerted cooling of the reaction mixture in air and in water: the workup.
[0]: The temperature of the reaction mixture was exclusively monitored by measuring the outside surface of the flask using an IR
thermometer. Hence, the reported values are crude approximations.
[1]: The duration of an external activity.
[2]: Adding an anti-foaming product containing emulsified silicone (for distilling spirits) crossed my mind since I had it on hand,
but I refrained as I wasn't absolutely certain of its compatibility; such a strategy seems worth exploring in the future, if need be.
[3]: The foaming was still taking place three hours into this period of six hours, but had completely subsided during the second
half.
[4]: 23.27 grams of distillate had been collected overnight/in total.
[5]: The escaping gas was most likely nitrogen. Dozens of bubbles were observed until its evolution appeared to cease, long before
the heating did. Volumetric measurement of this gas should provide some worthwhile insight relating to the overall progression of the reaction.
[Fig. 36] Initial portion of NaOH being stirred under MEG

[Fig. 37] Later state of the above mixture after adding more NaOH

[Fig. 38] Mixture following addition of 2,5-dimethoxyacetophenone

[Fig. 39] Reaction mixture after 15 minutes in heating mantle

[Fig. 40] After 30 minutes in heating mantle; beginning of reflux

[Fig. 41] Reaction mixture after refluxing for ~15 minutes

[Fig. 42] Threat of foaming over just as the 30-minute reflux is up

[Fig. 43] Reaction mixture cooling down

[Fig. 44] Equilibriation of foaming

[Fig. 45] Overnight distillate

[Fig. 46] Reoccurrence of foaming

[Fig. 47] Ostensible nitrogen exiting the apparatus

[Fig. 48] Post-reaction mixture cooling down

Work-up
600 mL of cold tap water was placed in a 1000 mL beaker, and the remaining cooled reaction mixture was poured in, followed by the pooled distillates
(50 mL). The resulting mixture was stirred for a while and suction filtered into a separatory funnel through cotton wool. The filter was rinsed with
several small portions of C7H16 (totaling 50 mL). After shaking the funnel and isolating the organic layer of rinses, two more
50 mL portions of heptane were used to extract the aqueous mixture, and the combined organic extracts were washed with a single 25 mL portion of
water, which was deposited in the aqueous partition. The heptane was removed via distillation at a reduced pressure, leaving 11.69 grams of a yellow
liquid with the viscosity of water and a strong, remarkably natural[1] carrot aroma with a hint of menthol. An exhaustive steam
distillation (which took about three hours) gave 11.31 g (83.8%) of a slightly volatile,[2] perfectly colorless, dense and immiscible
liquid phase via organic extraction of the distillate (140–150 mL) using three portions of DCM (10, 5 and 5 mL).
The aqueous partition was made acidic by addition of 33% HCl (45 g)[3] and extracted using three portions of DCM (30, 20, and 20 grams).
The pooled extracts were treated with saturated aqueous NaHCO3 (20 g of ~10% soln.) prior to distilling off most of the solvent. After
further evaporation on a watch glass, 2.20 g of an amber residue was obtained, which had the viscosity of sulfuric acid and a pleasant, sweet and
phenolic carrot aroma. This was stored on the watch glass, at 24°C, for 22 days, during which time it had darkened further and lost ~8% of its mass
(down to 2.02 g), before steam distilling it to collect 0.85 grams of a nearly colorless material, with properties[4] as before, floating
on ~65 mL of distillate.
[1]: A clean and distinctive raw carrot profile, accompanied by the earthy, moldy character that I associate with some carrot
farmers' cellar-like storage conditions. Interestingly, there's a clear resemblance between this and the comparatively subtle earthiness of the methyl
homolog, 2,5-dimethoxytoluene.
[2]: The material appears to evaporate; the rate of evaporation seems slower than that of 1,4-dimethoxybenzene or 1,4-benzoquinone,
but still significant.
[3]: After adding 40 g of the acid, a pH of 8–9 was measured.
[4]: Viscosity, odor, and gradual darkening on air exposure.
[Fig. 49] Pooled distillate and diluted post-reaction mixture

[Fig. 50] Crude phenolic side product <> Crude neutral main product

[Fig. 51] Steam distillation of desired product

[Fig. 52] Purified 2,5-dimethoxyethylbenzene

[Fig. 53] Steam distillation of phenolic side product

[e1] 2,5-dimethoxy-4-ethylbenzaldehyde
194.23 g/mol
314.40 g/mol (bisulfite adduct; potassium salt)
To formylate the finished benzene, I decided to attempt the Duff reaction, despite anecdotal evidence of impracticality,[1] as an
interesting way to gain some initial material to experiment with; the achievement of a worthwhile conversion seemed to require more specialized
methods. However, my expectations for the Duff were pleasantly exceeded, and, instead of attempting something different, I believe that a repeat
experiment is appropriate, somewhere down the road.
[1]: (Hyperlab) Pine_tar: Purified yield of 40.5%. https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
Experiment 1
To a magnetically stirred solution of 2,5-dimethoxyethylbenzene (5.00 g, 30 mmol) and hexamine (8.46 g, 60 mmol) in acetic acid (39.08 g), in a 100 mL
round boiling flask, there was added dropwise (in 40 minutes) a solution of 98% sulfuric acid (12.08 g, 121 mmol) in acetic acid (28.1 g) via a 250 mL
addition funnel. The resulting suspension of white precipitate was stirred for a further 10 minutes, after which the flask was placed into a 1000 mL
heating mantle, furnished with a Liebig condenser, and, with stirring, heated under reflux for 120 minutes. Next, the heating was discontinued, and
the mixture was allowed to cool below its boiling point before replacing the condenser with a short path still head that was used to remove acetic
acid (~33 g) by distilling the mixture for about 30 minutes. The Liebig was then reattached for the revival of reflux, and through it was added
1-butyl acetate (17.34 g), followed by a careful addition[1] of water (20 g) some five minutes later. The mixture was refluxed for a
further 100 minutes[2], allowed to cool, and stirred at room temperature for 15 more hours.
[1]: Water and 1-butyl acetate form a low-boiling azeotrope which can boil quite violently.
[2]: During this time, the color of the mixture shifted from a rather dark brownish red (fig. 58) to a considerably lighter shade of
reddish amber (fig. 59).
[Fig. 54] Solution of 2,5-dimethoxyethylbenzene and HMTA in AcOH

[Fig. 55] Above mixture following addition of sulfuric acid in AcOH

[Fig. 56] Beginning of reflux

[Fig. 57] Reaction mixture following addition of butyl acetate

[Fig. 58] Reaction mixture following addition of water

[Fig. 59] End of refluxing

Work-up
A small quantity of a pale, needle-like precipitate had formed in the mixture, and was redissolved by adding 10 g of water. The mixture was set aside
for about three hours before it was poured into a separatory funnel, rinsing the flask with some water and butyl acetate. The organic phase was
separated and combined with two 15-gram portions of BuOAc that had been used to further extract the aqueous mixture. The organic mixture was treated
with aqueous solutions of 25% NaCl (9 g), 10% NaHCO3 (9x10 g), and 25% NaCl again (5.5 g), followed by drying over 0.98 g of
MgSO4.
0.10 grams of the desiccated extract was placed on a watch glass to evaporate, leaving approximately 10 milligrams of a clear, yellow oil that
wouldn't crystallize spontaneously, even after tenacious rubbing with a glass rod. Interestingly, an immediate solidification to a waxy consistency
was observed when the oil came in contact with liquid water. A sample of this semisolid material was fought into a capillary tube with the help of a
second, thinner capillary tube, and a melting point range of 35.0–42.7°C was determined.
The remaining extract was concentrated by distilling off ~25 g of solvent and cooled back down to room temperature. A freshly prepared solution of
potassium metabisulfite (45.25 g) in water (100 g) was added to the vigorously stirred solution of the crude product. After about 20 minutes, the
mixture began rapidly thickening with the appearance of a solid adduct. The resulting porridge was stirred overnight (ca. 12 hours) and vacuum
filtered. The filter cake was rinsed with some butyl acetate and air-dried on a watch glass to yield 7.33 g of a nearly white solid.
The dried adduct was suspended in 155 mL of stirred water in a 250 mL beaker, and the mixture was basified to a pH of 10–11 by dropwise addition of
an aqueous 20% solution of NaOH (5 grams of the solution was used); the end point coincided with the evolution of a green hue. The suspension of
benzaldehyde was stirred for five minutes or so, and instead of vacuum filtering it, butyl acetate was added dropwise: 20.58 grams was added, with the
initial 2–3 grams being sufficient to dissolve everything, forming immiscible, milky droplets. The biphasic mixture was shaken in a separatory
funnel and left to stand for 26 minutes before collecting the organic layer and extracting the alkaline mixture with two more portions of the ester
(13 g, then 11 g). The combined organic extracts were washed with water (2.7 g) and 25% NaCl (10 g), followed by drying over 1.02 g of
MgSO4 until, after 30 minutes, the solution was clear.[1] The extract was gravity filtered through cotton into a 100 mL round
boiling flask along with a small portion of BuOAc used to rinse the flask and the desiccant, and most of the solvent was removed by distillation at
standard pressure.[2] The remaining solution was transferred onto a watch glass where the complete evaporation of volatiles gave 4.13 grams
(70.6%) of crunchy, unevenly discolored chunks of a glassy solid with a delightful odor of an obscure fruitiness coupled with the generic character of
freezer burn on a dairy-based ice cream; this had a melting point of 45.5–47.9°C (lit. 47–48°C). Recrystallization from an unrecorded excess of
isopropanol gave 3.45 g (59.0%) of a cleaner but still slightly yellow material which retained the aroma and melted at 46.5–47.6°C.[3]
[1]: The yellow discoloration that was present in the organic solution of the purified product seemed to be adsorbed (or otherwise
migrate) to the chunks of magnesium sulfate.
[2]: Something in the mixture seemed to exhibit an adverse reaction to the heating, producing a yellow discoloration. Possible
explanations include transformation of the benzaldehyde product as well as that of some impurity, stemming from the chemical extraction of the adduct
decomposition mixture. There will be further evidence to support this in the next update, but I'm going to state it right now: I think that mechanical
separation of the aldehyde following adduct decomposition is the way to go (where applicable).
[3]: Measured using a different thermocouple.
[Fig. 60] Organic solution of crude product <> Extracted RM and washings

[Fig. 61] Solidification of crude product following contact with water

[Fig. 62] Preliminary sample collected for melting point determination

[Fig. 63] Addition of bisulfite solution to crude aldehyde

[Fig. 64] Initial adduct formation

[Fig. 65] Above mixture prior to filtration

[Fig. 66] Isolated bisulfite adduct

[Fig. 67] Decomposition of adduct

[Fig. 68] Dissolution of purified benzaldehyde in minimal butyl acetate

[Fig. 69] Partitioning of decomposition mixture

[Fig. 70] Solution of purified product in BuOAc drying over MgSO4

[Fig. 71] Product obtained by distillation and evaporation of above solution

[Fig. 72] Recrystallized 2,5-dimethoxy-4-ethylbenzaldehyde

The next installment (4/5) will cover all of the sulfur chemistry, leaving the 2,4,6-trimethoxy motif for last. I aim to complete both before the year
is out, but it seems about as likely that I won't quite make it.
Either way, thank you for checking out this one!
[Edited on 26-11-2022 by Benignium]
|
|
|
arkoma
Redneck Overlord
Posts: 1761
Registered: 3-2-2014
Location: On a Big Blue Marble hurtling through space
Member Is Offline
Mood: украї́нська
|
|
| Quote: | | Either way, thank you for checking out this one! |
I thank YOU for posting the details of an obvious labor of love fueled by some kinda passion!!!
This was an epic thread when you started it Benignium, and now I fear I may run out of superlatives!!!
"We believe the knowledge and cultural heritage of mankind should be accessible to all people around the world, regardless of their wealth, social
status, nationality, citizenship, etc" z-lib
|
|
|
Pumukli
National Hazard
Posts: 705
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
I'm stunned, again, by your work, Benignium.
A lot of effort went into these synths (and obviously a lot of amateurish silliness too), but in the end you still have the results!
(Results, that most of us are probably a bit "positively envious" about. )
Keep up the hard work and keep us informed (and entertained) with the rest of the remaining preparations too! I swallowed your last post slowly, bit
by bit, during a several hours period, as if it was a small glass of the best beverage an amateur organic chemist could enjoy.
Your work is not only encouraging and entertaining but visually pleasing too!
[Edited on 26-11-2022 by Pumukli]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
For the acylation of p-dimethoxybenzene, I think you could probably use Ac2O with a Brønsted acid catalyst, e.g.:
https://www.sciencedirect.com/science/article/pii/S138111690...
Heteropolyacids are a lot of work but I think you can achieve similar results with H2SO4 or TsOH or something. Yields are already pretty good so won't
get much better, but a cleaner reaction and no noxious AcCl would be nice.
Your Duff results are consistent with my understanding that the Duff on p-dimethoxybenzene is not so great but when there is an activating
4-substitution, even alkyl, the yield is significantly improved.
Overall, really great work, and nice pictures.
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
arkoma - The thought of you, out there, supporting me with such vigor is an enduring source of improvement to my self-worth and
morale.
Pumukli - Your feedback is oozing a palpable sincerity — it is truly incredible. Thank you!
clearly_not_atara - Thank you for your support and the astute intellectual contributions that come with it!
|
|
|
tyro
Harmless
Posts: 31
Registered: 22-12-2021
Member Is Offline
|
|
| Quote: |
The methylation of 4-methoxyphenol seems to be about as pleasant as methylations get: the phenol is readily converted, and not terribly fussy when it
comes to oxidation, allowing for good yields of a remarkably fragrant[1] product, and doubtlessly the most satisfying substance that I've steam
distilled to date.
[1]: The odor is quite unique, but there's something very familiar about it. I suspect that much of this familiarity stems from its resemblance to
some species of clover that was (or were) abundantly present in my childhood.
|
The steam distillation of 1,4-dimethoxybenzene is incredibly satisfying, isn't it? I ran a few rounds of synthesis on this compound two or so years
ago, starting from hydroquinone and dimethylcarbonate. The product also tended to freeze in the condenser. I ended up having to stop the water pump a
few times to let the steam melt the mass which was threatening to clog up. And the smell? Beautifully pungent, definitely filled the working space and
then some... After smelling it in this context, I started to notice notes of it in cosmetics and perfumes.
Thanks for the incredible thread here, and all of your other fantastic contributions. It's really been a joy to read.
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
Oh yes — I wasn't quick enough (or my mantle wasn't hot enough) to avert the blockage by stalling the coolant, but the fix was thankfully still
simple enough to not devalue the experience.
Beautifully pungent is a great way of putting it; I wonder if there are those who genuinely dislike the scent. Overall, I've found the phenols and
their methyl ethers thus far to exhibit perfume-worthy fragrances with an astonishing frequency — all the while offering fascinating insight into
the relations of molecular structure and our olfactory perception.
I'd love to read more on your experiences with dimethyl carbonate!
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
Maximum effort.
The 2,5-dimethoxy-4-(alkylthio)benzaldehydes
212.27 g/mol (R = methyl)
226.30 g/mol (R = ethyl)
240.32 g/mol (R = 1-propyl)

Historically, the formation of the 4-alkylthio substituent has been regarded as a pick-and-mix of excessively
difficult, prohibitively hazardous, and/or atrociously malodorous procedures by the amateur chemist, leaving most with no other option than to give up
the chase. At the same time, however, the technology to overcome these barriers has been out there, waiting to be commonly accepted and
adopted through experimental rediscovery. [1][2] Such an initiative was eventually
taken by the Sciencemadness user Ullmann, whose absolute treasure chest of a thread [3]
I would have initially overlooked, were it not for a chance encounter with a more recent thread [4] in which another Sciencemadness user, turd, has done an outstanding job in presenting the core issues, and the ways in
which Alexander Shulgin, Ullmann, and later they themselves tackled them. Without these contributions, my own efforts in this area would exist only in
my dreams.
This post comprises my darnedest efforts to compile the entire diverging, 11-step sulfur chemistry portion of the overall endeavor
into a concise narrative whose progression remains faithful to the staged approach, according to which the actual events unfolded and interconnected
observations were made: in brief, a thiophenolic precursor was prepared, and divided into three portions (excluding an analytical sample), two of
which were selectively alkylated at the sulfur prior to stages of jugate permethylation and formylation with the third portion.
Even though some of the materials did sport a foul odor — mostly when impure — none were anywhere near as bad in terms of
volatility or detection threshold, or even in the qualitative sense, as the simple thiols (encountered in alternative approaches) are purported to be.
Honorable mentions should, however, be given out to a very impure sample of 2,5-dimethoxy-4-(methylthio)benzaldehyde, as well as a supposedly
microbial smell which haunted the immediate vicinity of the sink where glassware was cleaned; shortly after trace amounts of several of the
organosulfur materials were flushed down the drain, the exact same malodor would appear, and linger for a few days. Previously unknown, I would
describe the aroma as being reminiscent of decaying organic matter, and decidedly sewer-like. Interestingly, although the phenomenon certainly seemed
to coincide with the handling of everything from the thiophenol all the way to the methoxybenzenes (at least), disposal of several milligrams of the
five-month-old thiophenol down the same (interveningly inactive) sink caused no odors whatsoever.
Finally, this sequence of syntheses spans the breakage of a K-type thermometer and two thermocouples, which (along with some strange
values) is why, in December, I redetermined several of the melting point values — while also determining a few missing ones. This was done in one
session, using a thermocouple whose readings are reliable, albeit depressed by ~1°C (depending on the temperature range). Although the materials had
been stored for about 4–5 months (in airtight containers, at room temperature, and mostly protected from UV), they didn't appear to have degraded
significantly: only some rather superficial darkening was observed on the sample of propylthiohydroquinone. The verified values are given in
parentheses after the original values (not to be confused with literature values), and may be narrower due to a slower incrementation of temperature.
[1]: Lau & Kestner: Synthesis of 5-hydroxy-1,3-benzoxathiol-2-ones. https://doi.org/10.1021/jo01276a025
[2]: PiHKAL: #167 4T-MMDA-2. http://isomerdesign.com/PiHKAL/read.php?id=167
[3]: (Sciencemadness) Thread by Ullmann. https://www.sciencemadness.org/whisper/viewthread.php?tid=11...
[4]: (Sciencemadness) Thread by turd. https://www.sciencemadness.org/whisper/viewthread.php?tid=62...
Chapter I
THIOPHENOL
[a2] 1,4-benzoquinone
108.10 g/mol
Since the springtime experiments, I felt that I had unearthed sufficient evidence against water to revert back to using an alcoholic solvent. Because
I was able to conclude that my rather dilute hydrogen peroxide was definitely capable of producing benzoquinone, I prioritized improving the other
parameters over obtaining a higher concentration or seeking a different oxidizer altogether. Eventually, a striking experimental report by the
Sciencemadness user homeslice [1] convinced me to trial the profound modification of
bringing the complete reaction mixture to a boil for just 2–3 minutes. Even though this approach seemed to contradict my previous experimental
findings somewhat, it did prove exceedingly efficient.
[1]: (Sciencemadness) homeslice: Experimental report. https://www.sciencemadness.org/whisper/viewthread.php?tid=14...
Experiment 3 (-ish)
In a 1000 mL Erlenmeyer, hydroquinone (40.30 g, 366 mmol) and iodine (0.65 g, 2.6 mmol) were dissolved in isopropyl alcohol (150 mL) with mild heating
and magnetic stirring. There was then added, in one portion, 11.9% hydrogen peroxide (140 g, 489 mmol), after which the mixture was promptly heated to
its boiling point, maintaining reflux for two minutes under a 200 mm Liebig condenser [1] before
cessation of the heating. The mixture was stirred for another 10 minutes on the synchronously cooling hotplate prior to measuring its temperature at
~71°C (using IR); observing no more liberation of oxygen; and moving the flask onto a lab jack, where it was allowed to cool to 50°C over 20
minutes. Further cooling was effected by placing the flask in a cold water bath, where an abundant crystal formation was observed after 30 minutes.
[1]: The condenser was fitted with a U-bend (made up of adapters), whose purpose was to direct any spillage into an empty 500 mL
flask in the event of a runaway reaction. No boil-over occurred, but the vigorous evolution of oxygen did enable approximately a gram of
quinone-containing solvent vapor to make it past the condenser.
[Fig. 1] Iodine and hydroquinone under isopropanol
 [Fig. 2] Complete reaction mixture on the verge of boiling
[Fig. 2] Complete reaction mixture on the verge of boiling
 [Fig. 3] Above mixture about one minute later
[Fig. 3] Above mixture about one minute later
 [Fig. 4] Post-reaction mixture cooling down
[Fig. 4] Post-reaction mixture cooling down
 Work-up
Work-up
The crystal-laden flask was kept in the freezer for 90 minutes prior to vacuum filtering the mixture and rinsing the obtained solids with 5 g of
i-PrOH. Then, once the filter cake had been compressed and suctioned free of most of the alcohol, it was recrystallized from 35 g of
i-PrOH. The filtered crystals were placed (outside) in front of a fan in a crystallizing dish, where they were dried (with occasional mixing)
over about two hours. These crystals, while considerably cleaner, retained a rather dark overall shade of yellow, with a generous sprinkling of ones
that were outright reddish; a melting point of 115.5–117.7°C (lit. 115–116°C) was determined. [1] The material was placed in a brass mortar where it was triturated somewhat crudely under 35 g of i-PrOH.
Filtration of the mixture gave a slightly reddish liquor, and a high return [2] of a crystalline
powder which still appeared quite dirty. The solid was air-dried as before, and then recrystallized once more from 75 g of i-PrOH to little
apparent avail; after cooling the mixture in the refrigerator, vacuum filtering it, and washing the filter cake using 20 g of room-temperature
i-PrOH, the twice recrystallized, thrice air-dried yield of adamantly contaminated crystalline 1,4-benzoquinone was 31.14 g (78.7%).
[1]: The melting point test was expedited due to highly irritating fumes emanating from the heated capillary tube, leading to some
broadening of the experimental value.
[2]: No more than a gram of the quinone seemed to dissolve in the amount of (room-temperature) i-PrOH used.
[Fig. 5] Crystal formation in cooled post-reaction mixture
 [Fig. 6] Vacuum filtration to separate crude product
[Fig. 6] Vacuum filtration to separate crude product
 [Fig. 7] Separated crude product
[Fig. 7] Separated crude product
 [Fig. 8] First recrystallization; crystallized splatters of hot solution
[Fig. 8] First recrystallization; crystallized splatters of hot solution
 [Fig. 9] Recrystallized product
[Fig. 9] Recrystallized product
 [Fig. 10] Triturated product under fresh isopropanol
[Fig. 10] Triturated product under fresh isopropanol
 [Fig. 11] Second recrystallization
[Fig. 11] Second recrystallization
 [Fig. 12] Alternative perspective (after about a minute)
[Fig. 12] Alternative perspective (after about a minute)
 [b3] 5-hydroxy-1,3-benzoxathiol-2-one
[b3] 5-hydroxy-1,3-benzoxathiol-2-one
Also called: This
168.18 g/mol
Here's a very nifty little trick, and a key feature of this approach in terms of amateur-friendliness: when 1,4-benzoquinone is treated with
an excess of thiourea in the presence of a strong acid catalyst, a thiouronium salt is formed that can then rapidly cyclize on heating to give an
imine intermediate, which in turn hydrolyzes with the loss of ammonia to give the title compound. [1][2][3][4][5][6] Not only is this resulting heterocyclic intermediate valuable due to its convenient and high-yielding
formation and subsequent hydrolysis (c 3) — it also allows for the independent alkylation of each oxygen, paving the way to the
fascinating 2- and 5-monoethylated tweetios, like the 5-ethoxy-4-ethylthio-2-methoxyphenylethylamine. [6]
Going forward, it should be kept in mind that the thiophenol is remarkably sensitive to oxidation, and any handling in basic
conditions should be performed under an inert atmosphere, with care being taken to deoxygenate solvents beforehand.
[1]: Lau & Kestner: Synthesis of 5-hydroxy-1,3-benzoxathiol-2-ones. https://doi.org/10.1021/jo01276a025
[2]: PiHKAL: #167 4T-MMDA-2. http://isomerdesign.com/PiHKAL/read.php?id=167
[3]: (Sciencemadness) Thread by Ullmann. https://www.sciencemadness.org/whisper/viewthread.php?tid=11...
[4]: (Sciencemadness) Thread by turd. https://www.sciencemadness.org/whisper/viewthread.php?tid=62...
[5]: (Hyperlab) Obtaining mercaptohydroquinone. https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
[6]: (Hyperlab) miamiechin: 2C-T-2-5EtO. https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
Experiment 1
Onto a jointed 1000 mL Erlenmeyer flask were stacked, in order: a 200 mm Liebig condenser; a vertical vacuum distillation adapter with a length of
tubing leading to a dilute NaOH solution (gas scrubber); and a loosely stoppered 250 mL separating funnel. To a stirred solution of thiourea (23.28 g,
306 mmol) in approximately 2.5N hydrochloric acid (~244 mL, 557 mmol) [1] in the flask there was
added, at a rapid dropwise pace from the funnel on top, a solution of 1,4-benzoquinone (30.00 g, 278 mmol) in acetic acid (170 mL). The addition was
completed in 30 minutes, and it caused the temperature of the mixture to gradually rise to around 40°C. Once 15 minutes had passed since the end of
addition, the mixture had cooled to 37°C, and solids began to precipitate. The resultant, thick suspension was stirred for as long as it took to let
it cool down to as close to room temperature as it was going to get [2] before adding a further
portion of 33% HCl (18.50 g, 167 mmol) and then gently heating the mixture to reflux, where it was maintained until an hour had elapsed since it was
heated past 80°C. [3]
[0]: The temperatures were measured from exterior surfaces of the glassware using IR.
[1]: Prepared from 61.56 g of 33% HCl and 192 mL of water. Estimating a normality like this (as opposed to calculating it exactly via
titration of molarity) seems about as counter-intuitive as wearing sunglasses to bed, but there you go. In fact, full disclosure: none of my acids and
bases are properly titrated (nor have they been thus far), and claims like the above 557 mmol of HCl quite frankly do a much better job at
depicting my intentions with what I've been sold than they do the exact number of millimoles employed. That said, I'm not looking to advocate undue
laxness; what we have in titration is a fantastic opportunity to improve. First thing tomorrow.
[2]: This is ambiguously expressed as "After 90 minutes [added...]" in my notes, and based on the available documentation, it's hard
to tell whether this translates to 90 or 75 minutes following the end of addition. Either way, the mixture was slow to cool, and (IIRC) barely did so
past 30°C; I believe that there was some exotherm accompanying the solid formation, and that if frictional heat from the stirring was a thing, it was
in addition to a significant heat transfer from the active (i.e. perceivably warm) hotplate beneath.
[3]: The temperature of 80°C, while arbitrarily chosen for timing the reflux, was noteworthy for being roughly the point at which
bubbles began evolving on heating; likely signifying that a reaction was taking place.
[Fig. 13] Addition of benzoquinone solution to thiourea in hydrochloric acid
 [Fig. 14] Mixture following complete addition
[Fig. 14] Mixture following complete addition
 [Fig. 15] Alternative perspective (6500K lighting)
[Fig. 15] Alternative perspective (6500K lighting)
 [Fig. 16] Initial formation of solids
[Fig. 16] Initial formation of solids
 [Fig. 17] Suspension of supposed isothiouronium chloride
[Fig. 17] Suspension of supposed isothiouronium chloride
 [Fig. 18] Alternative perspective
[Fig. 18] Alternative perspective
 Work-up
Work-up
The post-reaction mixture was set aside to cool at room temperature. Crystals began forming from the solution at >60°C, and an impressive crop was
observed after seven more stationary hours. This was stored below 10°C for some hours prior to cooling to a temperature of 0–4°C, at which the
mixture was vacuum filtered. The unrinsed filter cake [1] was spread out over a coffee filter,
which was placed on several layers of paper towel. Air-dried, the crystals weighed 42.73 g (91.5%), melted at 177.7–179.8°C (lit.
170.5–172.5°C; [2] 175.5–176°C [3] ),
and had a rather pleasant, familiar odor with a low detection threshold. [4]
[1]: Not knowing much about the solubility of this product, and deeming it quite pure by appearance, I thought it best to not risk
rinsing it. In hindsight, it would have made a lot of sense to at least do a quick rinse with water; the solubility seems negligible, and the
obnoxious evaporation of acid fumes could have been mostly avoided.
[2]: PiHKAL: #167 4T-MMDA-2. http://isomerdesign.com/PiHKAL/read.php?id=167
[3]: SpectraBase: 1,3-BENZOXATHIOL-2-ONE, 5-HYDROXY-. https://spectrabase.com/spectrum/EjXXUEBshAz
[4]: Visually undetectable quantities on crudely wiped down surfaces could be smelled clearly. The
aroma bears an uncanny similarity to that of an odorous sample of impure crystalline MDMA which I encountered many years ago; whose origin was
completely unknown; and whose odor I have come to describe as resembling of root beer (which is entirely debatable). Having said all that, it's a
distinct possibility that, just as back then, the odor belongs to an impurity, and that none of it is inherent to the actual product. Interestingly, I
later found the odor of the crude 2-hydroxy-5-methoxyacetophenone (the phenolic side product of c 1) to exhibit a nearly identical
component.
[Fig. 19] Initial crystal formation
 [Fig. 20] Above mixture after seven hours at 24°C
[Fig. 20] Above mixture after seven hours at 24°C
 [Fig. 21] Obtained 5-hydroxy-1,3-benzoxathiol-2-one (in 6500K lighting)
[Fig. 21] Obtained 5-hydroxy-1,3-benzoxathiol-2-one (in 6500K lighting)
 [c3] 2,5-dihydroxythiophenol
[c3] 2,5-dihydroxythiophenol
Also called: Mercaptohydroquinone
142.18 g/mol
In this next step, the isolated intermediate is saponified using aqueous alkali to yield the desired thiophenol. While the obtained crude yield
appeared to be nearly quantitative, the initial melting point tests indicated a significant degree of impurity. It's possible that the conversion via
this described procedure may have been incomplete, as I certainly overestimated the purity of my NaOH. On top of this, there was observed what appears
to be a tendency to discolor and degrade on heating, which I suspect to be a characteristic of the product, and thus mostly independent of the initial
impurity. Prolonged storage in solution also seems like it could be problematic. All things considered, potential improvements include additional
base, thin-layer chromatography and, weirdly enough, not bothering with further purification.
[0]: For references, see the previous step.
Experiment 1
In a two-necked 1000 mL RBF, 5-hydroxy-1,3-benzoxathiol-2-one (40.01 g, 0.24 mol) was added through a 200 mm Liebig to a magnetically stirred,
deoxygenated [1] solution of NaOH (38.57 g, 0.96 mol) in water (320 mL), against a gentle
overpressure of argon. Maintaining the argon flow throughout, the mixture was heated under reflux for an hour and then moved to a water bath, where it
was cooled to ambient temperature prior to a colorful [2] acidification via dropwise addition of
33% hydrochloric acid (137.20 g, 1.24 mol).
[1]: Deoxygenation was performed by pulling an aspirator vacuum over the mixture several times, each time normalizing the pressure
using argon: first for a period of 5 minutes, and then three more times for as long as it took to reach the final depth of vacuum.
[2]: Green → gray → red amber (with initial foaming) → green → gray → nearly white with a green tinge, and opaque from
precipitation and effervescence.
[Fig. 22] Dissolution of starting ester in aqueous alkali
 [Fig. 23] Above mixture after about two minutes
[Fig. 23] Above mixture after about two minutes
 [Fig. 24] "Red amber" stage of post-reaction acidification
[Fig. 24] "Red amber" stage of post-reaction acidification
 [Fig. 25] Acidified mixture
[Fig. 25] Acidified mixture
 Work-up
Work-up
The acidified suspension was subjected to three vacuum-argon cycles, each giving rise (and fall) to a transient, pastry-like crust over an abundant
liberation of residual (hydrogen sulfide-smelling) gas. The mixture was then vacuum filtered to obtain a portion of solid material with a dirty white
color and a melting point of 109–116°C (lit. 118–119°C [1] ). Extraction of the filtrate
using ethyl acetate, followed by evaporation to dryness, gave a comparable amount of solid which was visibly cleaner and melted at 118–120°C. The
portions were combined (thoughtlessly, before knowing their melting points) for a crude yield of 33.19 g (98.1%).
Recrystallization of the crude product was explored. An unrecorded portion (presumably everything) was dissolved in 60 g of boiling ethyl acetate, and
20 g of heptane was added with the intention of lowering its solubility, but the liquids were immiscible. An initial crop of finer off-white crystals
(12.8 g, MP ~114°C) was obtained, followed by a second crop (10.9 g) of larger, more distinct crystals with a green hue from concentration of the
filtrate through distillation.
Evaporation of the concentrated mother liquor gave about six grams of a gooey, green, crystalline mass, from whence an extraction of product was
attempted with two portions of boiling toluene (25, then 21 g) that were decanted off of a sunken brown oil. The combined extracts were heated
together to redissolve everything. On cooling, a light-brown oil separated before the formation of crystals. A gram of n-PrOH was added in an
attempt to keep more of the oil in solution; the oil temporarily dissolved, but reappeared at a lower temperature, coinciding with crystal formation
and prompting the addition of another gram of n-PrOH. The mixture was heated to redissolve the crystals, and on cooling, the oil initially
remained in solution while crystals formed. At 24°C, however, some oil had still separated. The mixture was placed in a freezer and the cold liquor
was decanted off. Residual oil and solvent were removed by pressing the solids between layers of paper towel before air-drying them to obtain 2.57 g
of cream-colored crystals which melted at about 106°C.
The 10.9 g portion was recrystallized from 32 grams of aqueous 10% methanol to yield 8.09 g of crystals (with a melting point of ~117°C) via vacuum
filtration of the refrigerated (<10°C) mixture. The mother liquor was used to dissolve the residue from evaporation of the above toluene, as well
as the cream-colored crystals obtained from it, and the solution was allowed to slowly evaporate from a cling-film-covered beaker for about seven
weeks; the remaining liquid was then pipetted off, leaving yellow, crystalline solids which initially smelled like renally excreted asparagus
metabolites (if you know, you know), until they were completely air-dried and odorless, and weighed 3.11 g. A melting point range of 178–191°C
(180–188°C) was determined. [2]
The final yield of a uniformly off-white mixture of recrystallized materials seems to have been 23.58 g (69.7%). [3] Only the much later verified melting point value exists (110–117°C). The fresh material had a very slight green tinge,
which seemed to give way to a beige hue with time. I would describe the odor as a faint, sulfury sourness, with some of the root beer of the
ester precursor sticking through.
[1]: PiHKAL: #39 2C-T. https://isomerdesign.com/PiHKAL/read.php?id=39
[2]: At ~120°C, the material acquired a waxy appearance, as if moistened by some trace amount of
melted mercaptohydroquinone. As the sample melted, there was some simultaneous decomposition (i.e. reddening until orange), and a small amount of pale
solid material remained on the bottom. Interestingly, the previously mentioned "root beer" smell was distinctly emitted by the melted sample; this
could imply an incomplete saponification.
[3]: 19.22 g of the purified product was used and I still have 4.36 g left. This means that 2.69 g isn't accounted for in the notes,
and was most likely obtained from two initial small-scale recrystallization attempts from aqueous methanol, whose details are likewise missing — the
material should then originate from either the initial 33.19 g or [an unrecorded deduction that is missing from] the 12.8 g which was obtained from
the EtOAc recrystallization.
[Fig. 26] Rise of "pastry-like crust" under reduced pressure
 [Fig. 27] Combined fractions of crude product
[Fig. 27] Combined fractions of crude product
 [Fig. 28] Trial recrystallization from aqueous methanol
[Fig. 28] Trial recrystallization from aqueous methanol
 [Fig. 29] Combined impure fractions after seven weeks in beaker
[Fig. 29] Combined impure fractions after seven weeks in beaker
 [Fig. 30] Crystals from (the bottom of) above mixture
[Fig. 30] Crystals from (the bottom of) above mixture
 [Fig. 31] Purified material used in subsequent syntheses (surplus)
[Fig. 31] Purified material used in subsequent syntheses (surplus)

Chapter II
S-ALKYLATION
Another thoroughly fascinating feature of this approach is the exploitation of the differences between oxygen and sulfur as they relate to the atoms
being stuck on a benzene ring. A base, such as KOH, deprotonates a thiophenol in preference to a phenol due to the thiophenol being more acidic. In a
similar manner, an electrophile which is not particularly aggressive, like the alkyl bromides employed here, will have an overwhelmingly consequential
preference for a thiophenolate, which in turn is more nucleophilic than a deprotonated phenol. Therefore, what ends up happening, in this context, is
that even if one were to use more base than what is required to deprotonate the thiophenol — and even if there was a significant excess of the more
electrophilic ethyl bromide present — the immediate end result would be similar: a virtually exclusive and exhaustive alkylation of the
thiol. [1][2]
In light of the above, it is then clear to see that using more of each alkyl bromide (and quite possibly the base as well) would more
than likely have improved the outcome of these experiments; unreacted starting material most definitely contaminated both of the products, and the
same knowledge which could have been used to largely avoid the resultant issues was used to fix them in a bit of a leap of faith, whereby bicarbonate
was used to remove the (presumed) thiophenolic impurity in open air.
Briefly, the alkyl bromides used in these reactions were prepared via dropwise addition of sulfuric acid to a cooled mixture of KBr,
water and alcohol; refluxing the mixture; distilling the product from a hot water bath; washing the product with water, aqueous bicarbonate and brine;
and drying over MgSO 4.
[1]: (Sciencemadness) Thread by Ullmann. https://www.sciencemadness.org/whisper/viewthread.php?tid=11...
[2]: (Sciencemadness) Thread by turd. https://www.sciencemadness.org/whisper/viewthread.php?tid=62...
[d4] Propylthiohydroquinone
Also called: Hydroquinonyl n-propyl sulfide ( probably)
184.26 g/mol
Experiment 1
In a beaker, potassium hydroxide (3.13 g, 56 mmol) was dissolved in methanol (50 mL) with magnetic stirring, and the resulting rather polluted
solution [1] was gravity filtered into a double-necked 250 mL round boiling flask through a tiny
swab of cotton wool. With the flask set up in a water bath, [2] a dual-port gas adapter was
attached to the side neck, and used to connect both an argon bottle and a water aspirator; the vertical neck was stoppered. The alcoholic base was
then brought to a boil by pulling a vacuum which was immediately counteracted with an influx of argon to the point of (with help) lifting the stopper;
this was repeated two more times prior to replacing the stopper with a powder funnel and slowly adding 2,5-dihydroxythiophenol (7.11 g, 50 mmol)
through the sustained argon outflow. The mixture was allowed a moment of idle stirring before 1-propyl bromide (6.47 g, 53 mmol) was added in four
dropperfuls over about two minutes. Finally, the argon was terminated; the vertical neck was stoppered; and the mixture was stirred for 135 minutes.
[1]: The KOH (fig. 32) was green and contained some insoluble debris.
[2]: Both S-alkylations were performed in a ≤24°C water bath due to the unknown extent of potential exotherm. Aside from
inhibiting the volatilization of the alkyl bromides somewhat, no apparent benefit was had from doing this.
[Fig. 32] Technical grade potassium hydroxide
 [Fig. 33] Methanolic solution of KOH and mercaptohydroquinone
[Fig. 33] Methanolic solution of KOH and mercaptohydroquinone
 [Fig. 34] Reaction mixture within four minutes of 1-PrBr addition
[Fig. 34] Reaction mixture within four minutes of 1-PrBr addition
 [Fig. 35] Discoloration, likely caused by lingering atmospheric oxygen
[Fig. 35] Discoloration, likely caused by lingering atmospheric oxygen
 [Fig. 36] Post-reaction mixture
[Fig. 36] Post-reaction mixture
 Work-up
Work-up
The methanol was vacuum-boiled twice as before, and acidified (under argon) by adding a gram of 33% HCl. The walls of the flask were washed down with
MeOH using a dropper, [1] and the mixture was vacuum filtered. The filtered, slightly off-white
solids were washed using 12.7 g of MeOH and air-dried to obtain 4.67 g of (mostly) KBr which exhibited a slight, uneven darkening on air exposure. The
filtrate was distilled under reduced pressure to concentrate it down to a volume of ~40 mL before evaporating the remaining methanol under a PC fan
and transferring the residue to a water-containing beaker in order to examine its solubility (or complete lack thereof). The water was removed by
pipetting and evaporation, leaving 8.3–8.6 g of a clear, honey-colored oil.
After a fruitless attempt to obtain crystals from dissolving exactly one gram of the crude product in heptanes (10.24 g) with a bit of propan-1-ol
(1.5 g) — and subsequently converting it into a tarry mess by adding water, distilling off the organic solvents with a bunch of said water, adding
sodium bicarbonate solution to the remainder (and the distillate) to see what would happen, and stirring the mixture in the open flask to see for how
long [2] — the remainder was allowed to remain in open air on a watch glass. Not much change was
observed after seven days, aside from some strangely isolated discoloration near the borders of the glass, and perhaps a shift to a greener hue which
wasn't apparent at the time (fig. 41).
Inspired by the result of the preceding initial bicarbonate treatment of the ethylthio derivative (see d 3), the entire quantity of material
was transferred to a beaker using 3.6 g of ethyl acetate, and 50 mL of a 5% sodium bicarbonate solution was added. The biphasic mixture was stirred
magnetically for 5 minutes and then poured into a 250 mL separatory funnel. More EtOAc was added until, after adding ~12 g with occasional swirling,
the entire organic phase had risen on top of the aqueous one. The aqueous layer was drained into a beaker, where it was further extracted by stirring
with a 10 g portion of the EtOAc prior to being separated and acidified with 3.5 g of 33% HCl. The combined organic phases were washed with 40 g of
25% NaCl, and acidified with 0.78 g of 33% HCl. The acid-treated organic layer was gravity filtered through cotton and distilled to remove the bulk of
the solvent. The concentrated solution was poured into a shallow borosilicate dish where it solidified, affording 7.32 g (79.5%) of a cream-colored,
crystalline solid with a melting point of 69–72°C (70.4–71.4°C); this had a faint, fruity odor with hints of rubber and dehydrated onion.
Nothing was extracted from the aqueous portion.
[1]: The intention was to minimize discoloration from any residual deprotonated material getting oxidized before assimilating to the
acidified portion.
[2]: There was an immediate, progressive darkening which resulted in a nearly black mixture overnight (fig. 39). I don't have an
explanation for why this happened; the baking soda was reasonably fresh and appropriately stored, and I'm quite certain that the distilled mixture had
completely cooled beforehand.
[Fig. 37] Acidic vacuum filtrate and separated solids
 [Fig. 38] Crude product (mostly) under water
[Fig. 38] Crude product (mostly) under water
 [Fig. 39] Oxidative degradation of one-gram sample <> Also distillate
[Fig. 39] Oxidative degradation of one-gram sample <> Also distillate
 [Fig. 40] Freshly isolated crude product
[Fig. 40] Freshly isolated crude product
 [Fig. 41] Crude product after six days in open air
[Fig. 41] Crude product after six days in open air
 [Fig. 42] Above material dissolved in butyl acetate
[Fig. 42] Above material dissolved in butyl acetate
 [Fig. 43] Above solution being stirred with aqueous sodium bicarbonate
[Fig. 43] Above solution being stirred with aqueous sodium bicarbonate
 [Fig. 44] Purified propylthiohydroquinone
[Fig. 44] Purified propylthiohydroquinone
 [d3] Ethylthiohydroquinone
[d3] Ethylthiohydroquinone
Also called: 2-(ethylsulfanyl)benzene-1,4-diol
170.23 g/mol
On its surface, the ethylation appears identical to the previous propylation. However, there are a couple of considerations which were not taken into
account in performing this next experiment: firstly, ethyl bromide needs comparatively little provocation to peace out prematurely due to its
remarkably high vapor pressure; and lastly, it would then follow that continuously pumping an inert gas through the system which contains ethyl
bromide is strictly inadvisable — that is, unless there was an actual need to do so, as well as an excess of the bromide which was sufficient to
compensate. Which there wasn't.
Experiment 1
A solution of KOH (3.13 g, 56 mmol) in MeOH (50 mL) was gravity filtered into a two-necked 250 mL reaction flask, which was positioned in a cool water
bath and connected to argon and vacuum lines via a dual-port gas adapter on the angled neck. Four cycles of deoxygenation were effected in the same
manner as in d 4 prior to adding the thiophenol (7.11 g, 50 mmol), followed by EtBr (5.80 g, 53 mmol), and stoppering the vertical neck —
this time maintaining a slight, continuous efflux of argon into the aspirator reservoir while the mixture was stirred for two hours.
Work-up
To acidify the mixture, a gram of 33% HCl was added, followed by an additional portion of 10 drops. [1] The mixture was vacuum filtered to separate 3.77 g of KBr with an air-sensitive whiteness, and the bulk of the MeOH in
the filtrate was removed under vacuum before an exhaustive evaporation to leave a dark yellow oil.
In a bid to remove any unreacted thiophenol, the crude product was treated with bicarbonate. Using an unrecorded amount of ethyl acetate, the crude
product was transferred to a beaker, where it was magnetically stirred with 50 mL of 5% aqueous NaHCO 3 for a few minutes. The separated
organic solution was washed with 20 g of 25% NaCl, and then acidified using 6 g of 5.5% HCl prior to evaporating the solvent and thus obtaining 6.32 g
of a yellow oil. [2] No written descriptions of the odor seem to exist. [3] The bicarbonate partition was acidified using 2.38 g of 33% HCl and extracted using EtOAc to obtain 1.46 g of a
crystalline residue which was surprisingly light in color, [4] and practically odorless.
Eventually, after storing the product on a watch glass in open air for 19 days, [5] it was decided
that a second bicarbonate washing be carried out. The previously bicarbonate-treated product (6.25 g) was transferred to a beaker in 5.6 g of butyl
acetate. The solution was stirred with 50 mL of aqueous 5% NaHCO 3 for 12 minutes, after which the mixture was poured into a 250 mL
separatory funnel along with 8.4 g of additional BuOAc. Unlike before, the funnel was shaken, and quite a bit of discoloration immediately took place.
The aqueous layer was drained, acidified, and later extracted to obtain no residue. The organic layer was washed with 26 g of 25% NaCl, acidified with
1.06 g of 33% HCl, gravity filtered, and dried over 0.33 g of MgSO 4. To estimate the amount of product in the solution, 0.33 g (out of 20.5
g) was placed on a watch glass, leaving ~90 mg of residue after the evaporation of volatiles; this indicated ~5.59 g of product (65.7%).
Finally, ~11.7 g of the solvent was distilled off to leave a dark, viscous, reddish-brown oil in the flask. Prior to methylation, the flask was warmed
to ~50°C and swirled around while simultaneously purging it with argon to further remove residual volatiles.
[1]: Assuming a total volume of 0.5 mL and a density of 1.16 g/mL, this would be approximately 0.58 g.
[2]: A small sample of the oil was placed on a watch glass. Cooling in the freezer produced no crystals, but hardened the oil such
that its viscosity was comparable to something like barely malleable glass. The sample acquired a reddish discoloration on standing in open air (fig.
46); over the same span of time, the main portion remained unchanged.
[3]: I vaguely recall a modest, predominantly rubber-like smell, similar to what could be picked up from propylthiohydroquinone and
later its methyl ether — soft and sort of sweet, comparable to rubbers that are used in some worn and household items; footwear and
cleaning equipment, perhaps?
[4]: Part of my original assumption concerning the efficacy of a bicarbonate treatment was that any unreacted thiophenolate might
rapidly oxidize all the way to a hopeless tar following deprotonation. Moreover, I was quite certain that the tar would then immediately partition
into the organic solvent, tainting the hydroquinone. In a fascinating twist, the sort of dramatic oxidation that I was expecting didn't seem to take
place (unless the mixture is shaken with air in a funnel, apparently). Initially, I supposed that the presence of ethyl (and later even butyl) acetate
had a protective effect which allowed for the atmospheric isolation of 2,5-dihydroxythiophenol. However, when its melting point was finally determined
five months later; the isolated material was observed to gradually begin browning at ~120°C; partially melt into a chocolate-colored semisolid lump
at ~167°C; and then quite sharply blacken and liquefy completely at ~170°C — while the similarly stored actual thiophenol simply assumed
a chlorine-like color as it melted at 110–117°C. That said, I imagine that the impurity could still have been impure thiophenol, at the time.
[5]: On the first (or second) day, two tiny specks of the bicarbonate-extracted solid were placed in contact with the edges of the
puddle of crude product on the watch glass, with the intention of seeing whether they'd facilitate further solid formation or something else of the
sort. After seven days, pale streaks of ultra-fine particulate appeared to be spreading out from the solids (fig. 48), and at the end of the 19-day
period, the entire puddle was observed to have become opaque, with a ring of red discoloration around the edges (fig. 49); this was what prompted the
second bicarbonate treatment.
[Fig. 45] Impurity in BuOAc (aq. NaCl in the back) <> Product in BuOAc
 [Fig. 46] Discolored sample of product
[Fig. 46] Discolored sample of product
 [Fig. 47] Bicarbonate-extracted impurity
[Fig. 47] Bicarbonate-extracted impurity
 [Fig. 48] Streaks of particulate spreading over crude product
[Fig. 48] Streaks of particulate spreading over crude product
 [Fig. 49] Above sample after ~18 days in open air
[Fig. 49] Above sample after ~18 days in open air
 [Fig. 50] Aftermath of second bicarbonate treatment
[Fig. 50] Aftermath of second bicarbonate treatment

Chapter III
PERMETHYLATION
While the process of methylation itself is nothing out of the ordinary, the importance of protecting the hydroquinones from atmospheric oxygen is
highlighted, particularly in the first experiment, where the thiophenol is only now alkylated. To that end, I figured that I would try incorporating
acetone — which was previously used in the aqueous methylation of 4-methoxyphenol (b 1) without issues — the idea being that boiling it
under vacuum would drive out most of the dissolved oxygen. It has also been suggested that the presence of acetone can significantly inhibit the
saponification of dimethyl sulfate by the aqueous alkali, [1] which, if true (and applicable here),
would be a nice perk. Overall, the deoxygenation seemed to work well, but the yields were mediocre; this, along with the nearly identical dark brown
coloration that each of the post-reaction mixtures acquired on standing, seems to imply an incomplete conversion.
Because the territory felt somewhat more uncharted, the initial workup in particular involves a bunch of, as Cave Johnson
from Portal 2 might put it, "throwing science at the wall . . . to see what sticks". Out of the things that were attempted, the obvious
organic extraction and its treatment by repeated washing with aqueous alkali appeared to be the most effective, and I would consider these steps the
bare minimum in terms of isolation — kind of like I would for any of the other methoxybenzenes.
[1]: (The Hive) GC_MS: Acetone as co-solvent. https://the-hive.archive.erowid.org/forum/showflat/Cat-/Numb...
[d2] 1,4-dimethoxy-2-(methylthio)benzene
Also called: 2,5-dimethoxythioanisole
184.26 g/mol
Experiment 1
In a single-necked, 100 mL round boiling flask, [1] acetone (10 mL) was added to a solution of NaOH
(6.40 g, 160 mmol) in water (32 mL), and the magnetically stirred mixture was very briefly boiled three consecutive times by reducing the pressure and
immediately replacing the atmosphere with argon, whose continued outflow was then utilized to purge the subsequent addition of mercaptohydroquinone
(5.00 g, 35 mmol). The resultant solution was stirred for a couple of minutes, and a 250 mL separating funnel containing Me 2SO 4
(13.3 mL, 140 mmol) was attached, with simultaneous disconnection of the argon line to leave the apparatus open. The addition of DMS, initiated with a
rate of about 10 drops per minute, was completed in exactly one hour, with the reaction temperature hovering right around 37.4°C (per IR). The flask
was stoppered, and stirring was resumed for two hours before allowing the mixture to stand for about 52 hours at ambient outside temperatures
(13–23°C), pending workup.
[1]: The reaction flask was fitted with a vertical vacuum distillation adapter, which was connected to an argon bottle via its gas
port. A stopcocked 180° gas adapter was connected to a water aspirator, and attached to the female joint on the distillation adapter; once the vacuum
pump had served its function, the gas adapter was removed, and the starting material was added through the distillation adapter. This configuration
was utilized in all three of the methylations.
[Fig. 51] Reaction mixture prior to addition of DMS
 [Fig. 52] Reaction mixture after adding most DMS
[Fig. 52] Reaction mixture after adding most DMS
 Work-up
Work-up
The post-reaction mixture was poured into a 250 mL separatory funnel, diluted with 50 mL of water, and extracted using three portions of DCM (20, 5,
then 5 mL). The extracts were pooled and stripped of most solvent via distillation in a hot water bath, leaving approximately 5.8 g of a vibrant,
reddish-brown oil that had a fairly repulsive odor, reminiscent of dimethyl sulfide and sewage.
The usefulness of steam distillation was gauged by adding 32.67 grams of water to the crude oil and collecting ~10 mL of a biphasic distillate whose
bottom organic layer (<0.5 g) was pipetted onto a watch glass and evaporated along with two tiny portions of DCM used to crudely extract the
aqueous layer (for not much apparent gain). After a day of standing in open (24°C) air, the residue had shed most of its unpleasant cabbage-like
smell, and weighed ~90 mg. When no solidification was observed in over a week, the residue was placed in the freezer where it crystallized as a
pale-yellow mass which appeared to melt upon reaching ~18°C on standing at room temperature (lit. 33-34°C [1] ).
NaOH (0.98 g) was added to the remaining ~25 mL of crude product and water, and the mixture was stirred for 15 minutes or so prior to extraction with
three small portions of DCM; some color remained in the aqueous alkali. 20 mL of heptane was then added to the combined extracts in a 50 mL flask, and
the mixture was distilled to remove DCM, causing some dark, solid impurity to precipitate. Another 20 mL portion of heptane was added, and
distillation was resumed until ~15 mL of the mixture remained. This was then gravity filtered through cotton, rinsing the filter twice with fresh
heptane. A heavy, red oil separated as the mixture cooled, but was retained due to it potentially containing dissolved product; an addition of 10 mL
of toluene merged everything into a clear, amber solution.
0.57 g of finely ground activated charcoal was suspended in the solution at room temperature, and the mixture was then boiled for five minutes, cooled
in a water bath, and stirred for two more hours at room temperature. The charcoal was removed by vacuum filtration through qualitative filter paper,
affording a solution which was clearly less colored, but not dramatically so. A ~1% fraction of the solution was evaporated on a watch glass to obtain
~50 mg of an oil which crystallized in the freezer, but would melt at a significantly lower temperature (>0°C) than the previously steam distilled
sample.
Three more alkaline washes were performed using 10-gram portions of 10% NaOH; the first two washes assumed shades of yellow, and the third portion
remained colorless. A total of 3 g of methanol was then added to the third washing, until it caused some more of the discoloration to partition into
the aqueous layer. [2] The bright-orange organic solution was further shaken with a small portion
of water containing 0.52 g of 33% HCl, followed by an 11 mL portion of water with 0.51 g of dissolved potassium metabisulfite (both of which came out
colorless), before evaporation on a watch glass. The residual oil contained some trapped moisture, dust, and a fruit fly, which were separated: the
product wasn't soluble in plain heptane, but dissolved in a mixture containing 10% of toluene; this was then filtered through a ball of cotton.
Evaporation to a constant weight afforded 3.99 g of an amber oil with a faint, mildly unpleasant smell (4.08 g in total; ≤69.1%).
[1]: PiHKAL: #39 2C-T. https://isomerdesign.com/PiHKAL/read.php?id=39
[2]: This methanol-spiked washing was evaporated; the yellow, hygroscopic residue didn't seem to contain too much of the desired
methoxybenzene, and was discarded. All in all, the incorporation of methanol was not worthwhile.
[Fig. 53] DCM extract <> Aqueous partition
 [Fig. 54] Removal of DCM by distillation
[Fig. 54] Removal of DCM by distillation
 [Fig. 55] Activated charcoal <> Solution to be treated
[Fig. 55] Activated charcoal <> Solution to be treated
 [Fig. 56] Alternative perspective
[Fig. 56] Alternative perspective
 [Fig. 57] Charcoal-treated solution of product
[Fig. 57] Charcoal-treated solution of product
 [Fig. 58] Evaporated and refrigerated sample from above solution
[Fig. 58] Evaporated and refrigerated sample from above solution
 [Fig. 59] Solution of product over dilute HCl
[Fig. 59] Solution of product over dilute HCl
 [Fig. 60] Purified product with residual moisture and airborne impurities
[Fig. 60] Purified product with residual moisture and airborne impurities
 [e4] 1,4-dimethoxy-2-(propylthio)benzene
[e4] 1,4-dimethoxy-2-(propylthio)benzene
Also called: 1,4-dimethoxy-2-(propylsulfanyl)benzene
212.31 g/mol
Experiment 1
In a 100 mL reaction flask, acetone (16 mL) was added to a stirred solution of NaOH (4.84 g, 121 mmol) in water (35 mL), and — in the same manner as
before (d 2) — the mixture was deoxygenated prior to adding 1,4-dihydroxy-2-(propylthio)benzene (7.00 g, 38 mmol) and setting up a
dropwise addition of dimethyl sulfate (10.3 mL, 109 mmol). The entire addition took 30 minutes, with the reaction temperature remaining at ~36°C (IR)
throughout. The mixture was stirred for some time following the addition, [1] after which the flask
was stoppered and allowed to stand at ambient outside temperatures (above and below 20°C) for six days.
[1]: The exact time wasn't recorded, but most likely all three of the methylation experiments were stirred for two hours following
the end of addition.
[Fig. 61] Reaction mixture prior to addition of DMS
 [Fig. 62] Reaction mixture after adding most DMS
[Fig. 62] Reaction mixture after adding most DMS
 Work-up
Work-up
The mixture was diluted with 50 mL of water and extracted with four portions of DCM (20, 5, 5, and 5 mL). The bulk of the solvent was removed by
distillation, and the remaining oil was dissolved in 10 mL of heptanes. The addition of a 10 mL portion of 10% aqueous NaOH generated a small quantity
of dark sediment on stirring; the mixture was gravity filtered into a separatory funnel through cotton, and chased with a 10 mL portion of additional
heptane. The organic phase was retained, and washed with three more identical portions of alkali, followed by 10 mL of water, before it was drained
into a 50 mL flask. The separatory funnel was rinsed with a 10 mL portion of toluene which was also added to the flask. This nonpolar mixture was then
distilled atmospherically to the point of more dark sediment appearing, after which the distillation was resumed under vacuum until the precipitate
eventually redissolved in the residual oil. 10 mL of fresh heptane was then added, which once again precipitated the supposed impurity. The suspension
was refluxed for ~10 minutes with a gram of freshly ground activated charcoal. Vacuum filtration (whereby the glassware and separated charcoal were
rinsed with 2 mL of heptane) and evaporation of the filtrate to a constant weight gave 5.90 grams (≤73.2%) of a honey-colored oil with a mellow,
curiously zingy odor of sulfury rubber paired with some ambiguous middle ground of yellow onion and garlic.
Two weeks later, a ~0.80 g portion which wasn't used in the formylations was steam distilled. A coarse extraction of the collected slightly discolored
oil (from 200–250 mL of distillate) using DCM afforded, on evaporation, 0.69 g of an off-white, crystalline solid melting at 31.1–32.9°C
(32.1–33.7°C). The odor was unchanged.
[Fig. 63] Dark impurity resulting from treatment with base
 [Fig. 64] Steam distillation of unused product
[Fig. 64] Steam distillation of unused product
 [Fig. 65] Solid 1,4-dimethoxy-2-(propylthio)benzene
[Fig. 65] Solid 1,4-dimethoxy-2-(propylthio)benzene
 [e3] 1,4-dimethoxy-2-(ethylthio)benzene
[e3] 1,4-dimethoxy-2-(ethylthio)benzene
Also called: 2,5-dimethoxyphenyl ethyl sulfide
198.29 g/mol
The ethyl sulfide homolog was the last to be methylated. The digital notes on my phone seem to have encountered some deconstructive variation of
pocket dialing, whereby, in between the addition of dimethyl sulfate and the recorded yield, there is a section which simply states " 6 ".
Whatever was done was definitely very similar to the previous experiments: I recall reproducing the basic washing (also supported by fig. 68,
which I couldn't place anywhere else), and noting that the product is insoluble in heptanes; conversely, I'm convinced that I skipped the activated
charcoal treatment as well as attempting to steam distill the product.
Experiment 1
In a 100 mL reaction flask, a solution of NaOH (4.84 g, 121 mmol) in water (30 mL) was prepared. Acetone (10 mL) was then added prior to deoxygenation
of the mixture and addition of the crude 1,4-dihydroxy-2-(ethylthio)benzene (<6.25 g, <37 mmol) [1] via the established method; similarly, the argon cylinder was disconnected as part of initiating the subsequent dropwise
addition of DMS (10 mL, 105 mmol) which took 25 minutes to complete and generated a peak temperature of ~43°C (IR). After some additional stirring,
the mixture was moved to the side where it remained — stationary and outdoors — for no more than four days.
[1]: The lowest estimate for the amount of starting material used is 5.59 grams (see d 3 for details). Prior to its
addition, the viscous, oily material was diluted with a small portion of acetone, which was also used to sparingly rinse the adapter through which
said addition was made.
[Fig. 66] Reaction mixture prior to addition of DMS
 [Fig. 67] Reaction mixture at the end of addition
[Fig. 67] Reaction mixture at the end of addition
 Work-up
Work-up
The post-reaction mixture was diluted and extracted with several portions of DCM; the extract was washed with several portions of 10% NaOH solution,
before evaporating it to constant weight on a watch glass to obtain 4.85 g of a honey-colored oil which had a tangy, sickly-sweet fruit drop aroma
with a touch of rather off-putting, sulfury sourness. Depending on the actual amount of starting material used, the yield comes out at
(≤) 66.6–74.5%.
[Fig. 68] Separation of dark impurity (supposedly on treatment with base)
 [Fig. 69] All three dimethyl ethers, as utilized (Me \ Et / Pr)
[Fig. 69] All three dimethyl ethers, as utilized (Me \ Et / Pr)

Chapter IV
FORMYLATION
In recognition of my increasing vulnerability to failure, I began the formylations by dedicating a single gram of the most abundant intermediate to
doing a pilot experiment. At this point I had run out of ethyl acetate, and switching to the decidedly more stable butyl acetate elicited the idea of
incorporating it into the hydrolysis stage under reflux; this was done, and the crude yield from the pilot experiment ended up persuading me into
trying a similar procedure on the 2,5-dimethoxyethylbenzene right away, which reinforced the wishful impression that I just might be onto something.
The rest of the organosulfur compounds were then formylated — followed by the second portion of 2,5-dimethoxytoluene — and I have to admit: I
still haven't the faintest idea as to where I stand on the issue.
Here, the yields really weren't great, which begs the logical question "Why?" as well as perhaps an emotional exclamation mark or
two. Having pondered the matter, a slew of possible explanations has come to mind — none of which I can truly dispute, and many of which could have
been taking place in tandem. Through furious speculation, I found myself in a bit of a rabbit hole which I've attempted to narrate in the hopes that
it is of more interest to the chemist than it is to the psychologist.
Firstly, there's the Duff hydrolysis conundrum, elaborated on in the 4-Me post (1/5). Hypothetically, based on a statement
by the Hyperlab user miamiechin, [1] I might assume there to have been an initial yield of up to
~80% in each case, minus whatever percentage of impurity was present in the starting material. As an interesting side note, it initially seemed like
the crude yield of the propyl homolog from the pilot experiment could, by some metrics, [2] be
estimated to contain as little as ~10% of impurity, which would have put the yield at roughly 84%.
Building on these arcane assumptions, and the observed discrepancy between the yields of the two propyl experiments, it would appear
that a large chunk of each product might have been lost in the bisulfite purification. There's the distinct possibility of the formation and/or
precipitation of the bisulfite adduct of each aldehyde having been significantly incomplete, which seems to be supported by the observed decelerating
but seemingly everlasting precipitation of solids. Another correlation that stands out is how, instead of the usual dropwise addition of sodium
hydroxide, a concentrated sodium carbonate solution was used to decompose the obtained adduct from all but the pilot experiment; indeed, at least one
person has claimed that the use of (potassium) carbonate instead of hydroxide leads to lowered yields. [3] However, having tried the liquid–liquid extraction of each decomposition, the logistics for such a phenomenon seem
somewhat improbable to me, as I wouldn't expect any such loss to be soluble in the aqueous phase.
As my newest piece of conjecture, I now realize that there is one more thing which I have managed to repeatedly disregard as if it
kept curing scurvy: the acetic acid. In each experiment, 2.2–2.3 g of AcOH was used for every (supposed) millimole of starting material. Each
reaction mixture was then diluted to an arbitrary degree with water before a likewise arbitrary amount of butyl acetate was used to extract it. Beyond
this point, some interesting observations could be made: for instance, a mere 1.94 g of BuOAc per mmol (of starting material) was sufficient to
extract what amounted to a 69.6% purified yield of the methyl homolog, but subsequently washing the organic solution with aqueous bicarbonate would
then cause a significant quantity of the product to precipitate. Thus, it would appear that the amount of organically co-extracted acetic acid was
decreased via neutralization as well as partitioning to the aqueous phase, forcing the poorly BuOAc-soluble and clearly very hydrophobic aldehyde out
of solution. Similar correlations for the other two compounds aren't as conspicuous, but I believe that they're there. Mostly I'm just concerned that
I've insufficiently extracted the two seemingly less hydrophobic aldehydes from the acid-containing reaction mixture, but really the bedrock that I
keep unearthing here is that individual solubility of the [precursor/aldehyde/adduct] should be the constant main focus in deciding which variables to
adjust. At the end of this post, I've included a table that I drafted for reviewing and comparing the experimental parameters; with some tweaks, I
believe that it could be a great tool for planning and improving future Duff experiments as well.
Finally, I need to address the subject of melting points, as something strange seems to be going on with the separately S-alkylated
derivatives. Assuming that the melting point values given in PiHKAL are indeed correct, it would appear almost as if my ethylated and propylated
products had been switched at some point. However, I'm willing to risk the existential crisis of a lifetime in declaring that there is just no way
that that could have happened. And even then, the melting points wouldn't quite match — which shouldn't be an impurity issue considering the fairly
narrow ranges of 1.2°C and 0.9°C determined for the purest respective samples. Moreover, by (re-)verifying the melting point values in December, I
have completely ruled out the possibility of a thermometer malfunction. But if Shulgin didn't mix up his values; and if I didn't mix up my products
— what could have happened? Judging by the crude melting point of the pilot experiment, which at first seems to conform but then dramatically shifts
as the material is purified, it seems like the anomalous behavior could originate from the bisulfite purification process. But then, why wouldn't
something similar happen to the methyl homolog as well? It seems worth bringing up that Shulgin apparently used the Rieche (dichloromethyl methyl
ether/Lewis acid) to formylate the methylthio compound, and purified it via the bisulfite adduct; [4] while the ethyl and propyl derivatives were formed through the Vilsmeier, and simply recrystallized from methanol until
satisfactory [5][6] — with the latter apparently producing a perfect NMR spectrum. The best I
could do is some last-minute TLC, but characterization of any products from subsequent syntheses should in time supplement this nicely.
[1]: (Hyperlab) miamiechin: 70–80% yield. https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
[2]: SRS - Rule-of-thumb: 1% of foreign substance will result in a 0.5°C depression. https://www.thinksrs.com/downloads/pdfs/applicationnotes/MPP...
[3]: (Hyperlab) ramboTT_1: [7] (KOH vs. K2CO3). https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
[4]: PiHKAL: #39 2C-T. https://isomerdesign.com/PiHKAL/read.php?id=39
[5]: PiHKAL: #40 2C-T-2. https://isomerdesign.com/PiHKAL/read.php?id=40
[6]: PiHKAL: #43 2C-T-7. https://isomerdesign.com/PiHKAL/read.php?id=43
[f2] 2,5-dimethoxy-4-(propylthio)benzaldehyde
240.32 g/mol
360.49 g/mol (bisulfite adduct; potassium salt)
Experiment 1
In a 25 mL reaction flask, 1,4-dimethoxy-2-(propylthio)benzene (1.00 g, ≤4.7 mmol) and urotropine (1.32 g, 9.4 mmol) were mostly dissolved in
magnetically stirred acetic acid (6.20 g). To the mostly clear solution was then added a solution of 98% sulfuric acid (1.88 g, 18.8 mmol) in acetic
acid (4.12 g); the addition was performed dropwise, over 19 minutes, using a Pasteur pipette; and the ensuing cream-colored suspension was stirred for
five more minutes at room temperature. The flask was then fitted with a condenser, and its contents were brought up to a boil in a 1000 mL heating
mantle. After refluxing the mixture for 120 minutes, there was added butyl acetate (4.37 g) which separated a dense, dark oil. Once the heterogenous
mixture had been refluxed for another 80 minutes, water (5.03 g) was carefully added, and the heating was kept up for 60 more minutes before allowing
the mixture to cool to room temperature, at which it was then stirred for 13 hours.
[Fig. 70] Solution of substituted benzene and HMTA in AcOH
 [Fig. 71] Reaction mixture following addition of sulfuric acid
[Fig. 71] Reaction mixture following addition of sulfuric acid
 [Fig. 72] Reaction mixture prior to addition of butyl acetate
[Fig. 72] Reaction mixture prior to addition of butyl acetate
 [Fig. 73] Reaction mixture following addition of butyl acetate
[Fig. 73] Reaction mixture following addition of butyl acetate
 [Fig. 74] Reaction mixture following addition of water
[Fig. 74] Reaction mixture following addition of water
 Work-up
Work-up
The bilayered mixture was poured into a 250 mL separating funnel and shaken with additional water (25 mL) and BuOAc (10 g). Once partitioned, the
aqueous phase was extracted with two or three further 10 g portions of the ester, and the organic partitions were combined for treatments with water
(10 g), 10% sodium bicarbonate (3x10 g), and finally a second portion of water (10 g). The treated organic solution was gravity filtered into a 50 mL
flask through cotton (removing a small amount of dark sediment), and distilled until ~5 g of a yellow solution remained; this was evaporated to a
constant weight on a watch glass, yielding 1.06 g (<93.8%) of a waxy, yellow residue with a melting point of 71–77°C (lit. 76–77°C [1] ) and a small amount of (likely inorganic) impurity which didn't melt. Dissolution of the material in
11.8 g of methanol was incomplete, even with a gram of butyl acetate as a co-solvent, but the residual solids dissolved in the plain ester (1.88 g).
A solution of potassium metabisulfite (7.53 g) in water (16.8 g) was prepared. The methanolic solution of crude aldehyde was funneled into the stirred
bisulfite through cotton wool, and the mixture promptly thickened with solids that were pale apart from some dark orange bits here and there. The
previous 1.88 g portion of BuOAc was then added, which appeared to dissolve the colored impurity. After about 30 minutes, the mixture was vacuum
filtered, and the moist filter cake was strenuously rinsed with 8 g of fresh BuOAc before being placed on a watch glass to dry. The filtered mixture
continued to develop solids; a second filtration was performed after about an hour, followed by a third one on the next day. The filtrate was retained
for further observation.
In a beaker, the accumulated (partially dried) adduct was suspended in 50 mL of water with good stirring, and 2.35 g of a 20% NaOH solution was added
to raise the pH to 10–11, followed by 5 g of BuOAc used to dissolve the solid phase. The mixture was then transferred to a separating funnel, using
an additional 2.5 g of BuOAc to rinse the beaker. The phases were separated, and a further 0.44 g of the 20% NaOH was added to the aqueous
portion [2] prior to extracting it with two more five-gram portions of BuOAc. Once combined, the
organic extracts were washed with 10 g of 25% NaCl and a few grams of water. Most of the solvent was then distilled off, and as residual water was
removed with it, a small amount of supposed salt precipitated; the residual organic solution was drawn into a pipette through a swab of cotton and
deposited onto a watch glass to evaporate. Consequently, there was obtained 0.75 g (66.3%) of an odorless, slightly yellow, crystalline residue with a
melting point of 82.9–84.6°C.
[1]: PiHKAL: #43 2C-T-7. https://isomerdesign.com/PiHKAL/read.php?id=43
[2]: The presence of butyl acetate seemed to either buffer the pH down or distort the reaction
between the mixture and universal pH paper; a pH of ~9 was indicated, hence the addition.
[Fig. 75] Initial organic extract <> aqueous partition
 [Fig. 76] Residue from evaporation of above extract
[Fig. 76] Residue from evaporation of above extract
 [Fig. 77] Alternative perspective
[Fig. 77] Alternative perspective
 [Fig. 78] Formation of bisulfite adduct
[Fig. 78] Formation of bisulfite adduct
 [Fig. 79] Wet bisulfite adduct
[Fig. 79] Wet bisulfite adduct
 [Fig. 80] Bisulfite-purified product
[Fig. 80] Bisulfite-purified product
 [Fig. 81] Additional precipitation from retained bisulfite mixture
[Fig. 81] Additional precipitation from retained bisulfite mixture
 Experiment 2
Experiment 2
To a well-stirred solution of the benzene (4.04 g, ≤19 mmol) and HMTA (5.36 g, 38.1 mmol) in AcOH (25.55 g) in a 100 mL reaction flask, 98% sulfuric
acid (7.63 g, 76.2 mmol) in AcOH (18.13 g) was added dropwise (from a 250 mL addition funnel) over 22 minutes, causing the temperature of the mixture
to plateau at 31–32°C. Following completion, the thick suspension was stirred for seven more minutes at room temperature prior to being brought to
a reflux for 130 minutes. This was followed by one hour of stirring sans heating, after which there was added water (20.30 g) and BuOAc (14.68 g).
Stirring as such was continued for ~30 minutes, and the mixture was refluxed for an additional 40 minutes before allowing it to cool once more.
[Fig. 82] Reaction mixture being heated
 [Fig. 83] Above mixture 3 minutes later
[Fig. 83] Above mixture 3 minutes later
 Work-up (2)
Work-up (2)
After some 20 hours at room temperature, [1] the mixture was diluted with 15 g of water, and
partitioned. The aqueous portion was extracted twice with BuOAc (12, then 21 g), and the extracts were merged with the organic portion. This was
shaken twice with 25 g portions of water in a separatory funnel and then drained into a stirred suspension of NaHCO 3 (10 g) in 90 mL of
water; an additional three grams of NaHCO 3 was required to retain alkalinity in the aqueous phase (one gram followed by four half-gram
portions). The organic solution was separated and dried over 1.02 g of MgSO 4.
Omitting distillation [2] to remove excess solvent, a solution of potassium metabisulfite (35.52 g)
in water (78.63 g) was added to the dried organic solution with strong stirring. Solids began forming after about 10 minutes. After about 12 hours,
the mixture was vacuum filtered. The filter cake was rinsed with methanol, [3] which appeared to
give rise to further precipitation on mixing with the filtrate. [4] The cake was air-dried to a
constant weight of 3.44 g, and a second filtration of the mixture ~18 hours later gave 0.38 g of additional solids for a total of 3.82 g.
The twice harvested bisulfite mixture was partitioned. The organic portion was used to extract the remainder of the reaction mixture (where the two 25
g water washings had also been deposited). Another extraction was done using BuOAc (10 g), and the combined organic extracts were washed with water
(10 g), 25% NaCl (10 g), 10% NaHCO 3 (8x10 g), and 25% NaCl (10 g) again, before being dried over 1.25 g of MgSO 4 for ~20
minutes. Meanwhile, the aqueous bisulfite solution from before was replenished via dissolution of an additional 1.18 g of
K 2S 2O 5, and then combined with this new organic extract. The mixture was stirred for ~12 hours and filtered. The
solids were rinsed with MeOH (2 mL) and BuOAc (6 g). The air-dried solids (3.03 g) were merged with the previously obtained 3.82 grams.
In an attempt to ensure that all of the available benzaldehyde would be retained, the spent bilayered bisulfite mixture was combined with that of the
previous experiment. After 24 hours of standing, there had formed a bunch of additional solid material, which was separated by vacuum filtration and
rinsed with 2 mL of MeOH. This, in combination with a later 0.68 g portion of even more solid material (rinsed with BuOAc), weighed 3.85 g.
The two portions of ostensible adduct were processed forth separately, beginning with the latter. The solid was added to a ~26.2% solution of
Na 2CO 3 (17.49 g) in water (49.38 g), and the mixture was stirred for 15 minutes. There was then added 5 mL of butyl acetate,
which was noted to dissolve the solid phase sluggishly (in 10–20 seconds). The mixture was left to stand for a little over an hour before the phases
were separated and the aqueous portion was extracted with two 5 mL portions of BuOAc. The organic extracts were combined, dried over 0.31 g of
MgSO 4 and evaporated on a watch glass to yield 0.32 g of a crystalline residue with a melting point of 83.6–84.6°C.
The former 6.85 g portion was similarly decomposed in 80 g of the ~26.2% Na 2CO 3. The solid phase didn't seem to dissolve too
well, with most of it persisting after [probably 5–15 minutes] of being suspended in a stirred emulsion containing 10 mL of butyl acetate. Instead
of adding more BuOAc right away, the mixture was vacuum filtered. The filtered solids dissolved partially when rinsed with a 10 mL portion of BuOAc,
so an approach was chosen whereby the same biphasic mixture was repeatedly filtered through the solids into a separatory funnel, with BuOAc being
added incrementally until, after adding three more portions (10, 10 and 5 mL) for a total of 45 mL, only a few dozen crumbs remained. [5]
The liquid mixture was partitioned, and the organic solution was dried over 0.64 g of MgSO 4 for >24 hours. No longer omitting
distillation (due to forgetting about [2] ) the dried solution was distilled to remove the bulk of
the solvent prior to evaporation of the remainder on a watch glass to yield 2.38 g (52.0%) of a wintry, crystalline residue melting at 82.4–84.6°C.
A recrystallization of the combined product of both experiments (3.45 g) from ~19 mL of isopropanol gave 3.21 g (56.3%) of odorless, cream-colored
crystals with a melting point of 84.1–84.9°C (82.9–83.8°C).
[1]: It isn't specified whether the mixture was stirred or just stood there for ~20 hours.
[2]: Caution due to the observed heat-induced discoloration during the preceding workup of the 2,5-dimethoxy-4-ethylbenzaldehyde, as
well as a suspicious discoloration of the purified product from the pilot experiment.
[3]: The amount wasn't recorded; it was added in one portion using a 10 mL pipette, and was definitely no less than 2 mL. I seem to
recall using 6 mL but am way too uncertain to conclude thus.
[4]: This could mean an improvement to the rate and/or quantity of adduct formation, but it could also just be the precipitation of a
dissolved portion of the filter cake and/or some insolubilized undesired material. As mentioned previously, I believe that methanol generally has a
beneficial effect on the adduct formation through acting as a phase transfer catalyst (where applicable).
[5]: These crumbs were identified as mostly product by their mixed melting point. However, they were pending characterization long
enough to be excluded from the initial yield and its recrystallization, and, by extension, from the purified yield. I seem to have since misplaced the
crumbs, but I'd say there was probably no more than 100 mg (fig. 90).
[Fig. 84] Organic extract over aqueous bicarbonate suspension
 [Fig. 85] Treated extract being stirred with bisulfite solution
[Fig. 85] Treated extract being stirred with bisulfite solution
 [Fig. 86] Above mixture after 11 hours of stirring
[Fig. 86] Above mixture after 11 hours of stirring
 [Fig. 87] 3.85 g portion of adduct being added to aqueous alkali
[Fig. 87] 3.85 g portion of adduct being added to aqueous alkali
 [Fig. 88] Above mixture after 13 minutes of stirring
[Fig. 88] Above mixture after 13 minutes of stirring
 [Fig. 89] Above mixture following addition of butyl acetate
[Fig. 89] Above mixture following addition of butyl acetate
 [Fig. 90] Extracts from the two individual decompositions drying
[Fig. 90] Extracts from the two individual decompositions drying
 [Fig. 91] Undissolved crumbs of product on filter paper
[Fig. 91] Undissolved crumbs of product on filter paper
 [Fig. 92] Evaporation residue of bisulfite-purified product
[Fig. 92] Evaporation residue of bisulfite-purified product
 [f1] 2,5-dimethoxy-4-(ethylthio)benzaldehyde
[f1] 2,5-dimethoxy-4-(ethylthio)benzaldehyde
226.30 g/mol
346.47 g/mol (bisulfite adduct; potassium salt)
Experiment 1
1,4-dimethoxy-2-(ethylthio)benzene (4.60 g, ≤23.2 mmol) and HMTA (6.50 g, 46.4 mmol) were dissolved in AcOH (30.4 g) in a 100 mL reaction flask. To
the stirred solution was then added, dropwise, over 33 minutes, a solution of 98% H 2SO 4 (9.29 g, 92.7 mmol) in AcOH (22 g). On
completion, the mixture was refluxed for 90 minutes after which there was added (through the condenser) BuOAc (19.00 g), followed by a cautious
addition of water (10.40 g). After refluxing the reaction for another 70 minutes, it was allowed to cool for 20 minutes prior to adding a second
portion of water (10 mL). Stirring was then continued for ~19 hours at 24°C.
[Fig. 93] Addition of HMTA to substituted benzene in acetic acid
 [Fig. 94] Reaction mixture prior to heating
[Fig. 94] Reaction mixture prior to heating
 [Fig. 95] Reaction mixture approaching reflux
[Fig. 95] Reaction mixture approaching reflux
 [Fig. 96] Above mixture after 10 minutes
[Fig. 96] Above mixture after 10 minutes
 [Fig. 97] Mixture after 50 minutes of refluxing with water present
[Fig. 97] Mixture after 50 minutes of refluxing with water present
 [Fig. 98] Monophasic post-reaction mixture
[Fig. 98] Monophasic post-reaction mixture
 Work-up
Work-up
In a separatory funnel, the monophasic post-reaction mixture was shaken with 53 mL of water and 15 g of BuOAc, and the two layers were separated. The
aqueous portion was extracted twice using 10 g portions of BuOAc. Once combined, the organic portions were treated with 10% NaHCO 3 (4x20 g,
then 2x10 g) until a washing remained alkaline. The amber organic solution was dried overnight over 1.15g MgSO 4. A 0.17 g sample was
evaporated to dryness in a 10 mL beaker to obtain an unweighed, crystalline melting point sample with an unpleasant odor. [1]
A bisulfite solution (38.7 g of K 2S 2O 5 in 86.2 g of H 2O) was added to the stirred, gravity filtered
organic solution in a 250 mL flask, along with 10 mL of MeOH. In ~10 minutes, the mixture had turned into an unstirrable porridge. The flask was
swirled occasionally over the next 14 hours prior to vacuum filtering the mixture, washing the filter cake with 6 mL of MeOH followed by a total of 20
mL of BuOAc, and air-drying the obtained solids to a constant weight of 6.28 g.
Because the dried adduct had a rather dirty yellow color which wouldn't wash away on the filter, it was magnetically stirred with 20 mL of methanol in
a 50 mL flask for about two hours. Filtration and air-drying of the mixture afforded 4.40 g of a significantly less discolored solid. The filtrate was
combined with the separated aqueous portion of the previously used bisulfite mixture; [2] powerful
stirring of the mixture followed by vacuum filtration, and drying of the separated solids, gave 1.53 g of material which was likewise much less
discolored than what was had before.
A dark, crystallizing oily substance was observed to appear on top of the mixture of washings and extracted reaction mixture; a ~40 mL portion of the
distilled organic phase from the bisulfite mixture was used to re-extract the mixture. The extract was treated with two portions of 10%
NaHCO 3 (40 g, 30 g) followed by 10 g of 25% NaCl, and then stirred overnight with the pre-used aqueous bisulfite to receive 3.18 grams of
additional precipitate.
The accumulated adduct was added to 83 g of stirred ~26.2% sodium carbonate solution. After waiting 15 minutes, 10 mL of BuOAc was added, and the
mixture was stirred for 10 minutes. The solid phase dissolved partially. Two more five-milliliter portions of the ester were needed to obtain a clear
mixture; these were added 10 minutes apart. A small quantity of unidentified debris appeared at the liquid–liquid interface of the still mixture,
and was gravity filtered out. The organic phase was separated, and the aqueous phase was extracted twice more with 10 mL portions of BuOAc. The
combined organic extracts were dried over 0.76 g of MgSO 4 for ~24 hours, and concentrated through distillation under reduced pressure.
Following evaporation of the residual volatiles, there remained 2.62 g of slightly sticky, yellow crystals melting at 73.6–79.8°C (lit.
87–88°C [3] ). These were recrystallized from ~28 mL of isopropanol, giving 2.38 g (45.3%) of
crispy, odorless crystals which were lighter in color and had a melting point of 77.1–78.8°C (76.9–78.1°C).
[1]: For whatever reason, it seems that the melting point was never determined. But there is a picture of the sample (fig.
101).
[2]: In hindsight, it seems likely that much of the dissolved portion consisted of the adduct, and instead of just putting it back
(and probably hurting the yield in doing so), it would have been prudent to evaporate the MeOH and examine the residue.
[3]: PiHKAL: #40 2C-T-2. https://isomerdesign.com/PiHKAL/read.php?id=40
[Fig. 99] Aqueous portion of diluted reaction mixture <> Organic extract
 [Fig. 100] Washed extract <> Washings and above aqueous portion
[Fig. 100] Washed extract <> Washings and above aqueous portion
 [Fig. 101] Underutilized sample of impure product
[Fig. 101] Underutilized sample of impure product
 [Fig. 102] Treated extract being stirred with bisulfite solution
[Fig. 102] Treated extract being stirred with bisulfite solution
 [Fig. 103] Above mixture after 12 minutes of stirring
[Fig. 103] Above mixture after 12 minutes of stirring
 [Fig. 104] Dried bisulfite adduct
[Fig. 104] Dried bisulfite adduct
 [Fig. 105] Decomposition of adduct
[Fig. 105] Decomposition of adduct
 [Fig. 106] Above mixture after adding 20 mL of butyl acetate
[Fig. 106] Above mixture after adding 20 mL of butyl acetate
 [Fig. 107] Evaporation of extract from adduct decomposition
[Fig. 107] Evaporation of extract from adduct decomposition
 [Fig. 108] Alternative perspective (distorted white balance)
[Fig. 108] Alternative perspective (distorted white balance)
 [e2] 2,5-dimethoxy-4-(methylthio)benzaldehyde
[e2] 2,5-dimethoxy-4-(methylthio)benzaldehyde
212.27 g/mol
332.44 g/mol (bisulfite adduct; potassium salt)
Experiment 1
In a 100 mL reaction flask, there was prepared a solution of 2,5-dimethoxythioanisole (3.99 g, ≤21.7 mmol) and hexamine (6.08 g, 43.4 mmol) in AcOH
(29 g). A solution of 98% H 2SO 4 (8.68 g, 87.0 mmol) in AcOH (19 g) was then added — dropwise, and with strong stirring — in
30 minutes, during which the reaction mostly took place at ~31°C. Following completion, the mixture was stirred for an additional 10 minutes at
ambient 24°C prior to being refluxed for two hours. Water (10 g) and BuOAc (10 g) were then added, followed by another portion of water (10 g) after
45 more minutes of refluxing, at which point the heating was discontinued. After six hours of stirring, the mixture was warmed to 70°C (with a
short-lived intention of further refluxing) and allowed to cool again. The mixture was stirred for 17 more hours and allowed to stand for another two.
[Fig. 109] Solution of substituted benzene and HMTA in AcOH
 [Fig. 110] Reaction mixture following addition of sulfuric acid in AcOH
[Fig. 110] Reaction mixture following addition of sulfuric acid in AcOH
 [Fig. 111] Reaction mixture approaching reflux
[Fig. 111] Reaction mixture approaching reflux
 [Fig. 112] Reaction mixture after 110 minutes of reflux
[Fig. 112] Reaction mixture after 110 minutes of reflux
 [Fig. 113] Reaction mixture prior to second addition of water
[Fig. 113] Reaction mixture prior to second addition of water
 [Fig. 114] Monophasic post-reaction mixture
[Fig. 114] Monophasic post-reaction mixture
 Work-up
Work-up
The mixture was transferred to a separatory funnel where it was diluted with 50 g of water, causing a great deal of precipitation; the addition of 10
g of BuOAc redissolved the solid phase on shaking. The organic phase was collected and combined with two more ester extractions (12 g, 10 g), and the
resulting mixture was treated with two 50-gram portions of 10% NaHCO 3; this produced a pale precipitate [1] which required several iterative solvent additions to redissolve: in order, there was added, portionwise, BuOAc (12 mL),
DCM (1, then 2 mL), water (20 mL), and DCM (7, 10 and 5 mL). Finally, only a few solid particles remained, and the biphasic mixture was partitioned.
Treatment of the product-containing partition was resumed with a further 5 g of the aqueous bicarbonate, followed by 10 g of 25% NaCl. There was then
added 0.97 g of MgSO 4. [2]
The mixture was distilled to remove DCM, which was then used to further extract the remainder of the reaction mixture, where solids had now
formed. [3] The latter extract was concentrated and then evaporated to yield 440 mg of a vibrant,
reddish-orange residue with a rather vomit-inducing smell, reminiscent of the truly visionary delicacy known as stockfish; melting point 71–91°C.
To the former extract, there was added a bisulfite solution (36.51 g of K 2S 2O 5 in 81.01 g of H 2O) and 10
mL of methanol; the mixture almost immediately became unstirrably thick. Vacuum filtration of the mixture was performed a total of three times, as the
filtrate would thicken once more before finally producing a ~100 mg portion of additional solid. Thus, in total, there was obtained 8.74 g of dried
precipitate.
The obtained adduct was suspended in 85 g of ~26.2% Na 2CO 3, and the mixture was stirred for 15 minutes before adding 15 mL of
BuOAc, which resulted in no discernable dissolution but rather constituted a mesmerizing, doughy vortex with the hydrophobic benzaldehyde. When an
additional 15 mL portion of BuOAc (and 10 minutes of stirring on either side of it) made no apparent difference to the quantity of solids, the mixture
was vacuum filtered through qualitative filter paper, and the filter cake was rinsed with 100 mL of water to obtain, after air-drying, 3.01 g of a
pale, greenish-yellow solid exhibiting a slightly incomplete melting at 97.6–99.6°C (lit. 99–100°C [4] ). The organic portion of the filtrate was washed with the previous 100 mL portion of water, filtered, and dried over
0.39 g of MgSO 4; approximately two percent (0.51 g) of the solution was evaporated to obtain ~10 mg of a residue melting at 99.0–99.6°C.
Recrystallization was trialed by first gauging the solubility in 2-propanol: a 0.50 g portion of the solid material was dissolved in ~8 mL of the
alcohol, and the boiling mixture was gravity filtered through cotton to remove a small quantity of insolubles; by visually inspecting the crop of
crystals from cooling the liquor, the ratio of solvent was deemed apt. The rest of the solids were then dissolved in 32 g of 2-PrOH, gravity filtered,
and combined with the two fractions in alcohol and BuOAc. The mixture was distilled to remove alcohol [5] and placed in the freezer. Vacuum filtration of the <0°C mixture gave 3.20 g (69.6%) of nearly colorless but
deceivingly fluorescent, odorless crystals with a melting point of 101.0–101.5°C (96.2–96.9°C). [6]
[1]: This (along with the other instances of unexpected precipitation) was most likely the product, whose solubility in butyl acetate
seemed to be the lowest of the three homologs (in fact, of all of the benzaldehydes so far). In the notes, I haven't specified whether it was the
first or the second bicarbonate washing which caused the precipitation, and it could be that both portions ended up in the funnel at the same time
with all of the additional organic solvent, with the third five-gram portion being used to verify complete neutralization of the acid.
[2]: It is unclear if the solution was actually dried (and filtered) prior to the subsequent distillation; DCM is a considerably
better solvent for the product than BuOAc, ergo, its (sufficient) removal should precipitate the product — mixing it with the sulfate, if the
sulfate was present. In any case, it does seem like I made it to the bisulfite purification with no such incident.
[3]: Another detail was omitted in not noting whether the re-extracted mixture contained the previous bicarbonate washings; as I
recall, it did.
[4]: PiHKAL: #39 2C-T. https://isomerdesign.com/PiHKAL/read.php?id=39
[5]: The final volume of solvent was crudely estimated (after the fact) to be ~35 mL.
[6]: The verified value was obtained by placing the sample in the furnace whose temperature was already quite close to the melting
point; the abrupt heating seems to have been the cause of the slight depression.
[Fig. 115] Precipitation on rinsing reaction flask with water
 [Fig. 116] Precipitation on aqueous dilution of reaction mixture
[Fig. 116] Precipitation on aqueous dilution of reaction mixture
 [Fig. 117] Precipitation on washing extract with aqueous bicarbonate
[Fig. 117] Precipitation on washing extract with aqueous bicarbonate
 [Fig. 118] Spoonful of bisulfite adduct from the source
[Fig. 118] Spoonful of bisulfite adduct from the source
 [Fig. 119] Precipitation during secondary extraction of reaction mixture
[Fig. 119] Precipitation during secondary extraction of reaction mixture
 [Fig. 120] Secondary extract
[Fig. 120] Secondary extract
 [Fig. 121] Evaporation residue of above extract
[Fig. 121] Evaporation residue of above extract
 [Fig. 122] Dried bisulfite adduct
[Fig. 122] Dried bisulfite adduct
 [Fig. 123] Decomposition of adduct in aqueous sodium carbonate
[Fig. 123] Decomposition of adduct in aqueous sodium carbonate
 [Fig. 124] Water floating on aldehyde floating on water — under BuOAc
[Fig. 124] Water floating on aldehyde floating on water — under BuOAc
 [Fig. 125] Above mixture, vigorously stirred
[Fig. 125] Above mixture, vigorously stirred
 [Fig. 126] Solids from vacuum filtration of above mixture
[Fig. 126] Solids from vacuum filtration of above mixture
 [Fig. 127] Purified aldehyde under 2700K white LED (corrected WB)
[Fig. 127] Purified aldehyde under 2700K white LED (corrected WB)
 [Fig. 128] Purified aldehyde under UV-A
[Fig. 128] Purified aldehyde under UV-A
 [Fig. 129] Illumination: Indirect evening sunlight <> UV-A
[Fig. 129] Illumination: Indirect evening sunlight <> UV-A
 [Fig. 130] Table for reviewing experimental parameters
[Fig. 130] Table for reviewing experimental parameters
 Thin-layer chromatography
Thin-layer chromatography
To lessen the ambiguity of this post, I decided to squeeze in some TLC; despite some initial perplexity, useful results were obtained. Samples in the
approximate range of 3–6 milligrams were placed in test tubes, each containing a gram of isopropanol. Four identical plates (25 x 75 mm) were
prepared by applying a spot of each sample, which were labeled as A (Me), B (Et), and C (Pr). Each
of the plates was developed using a different mobile phase before being placed in chronological order and photographed under UV-A and UV-C lighting.
Mobile phases, from left to right:
[1] Toluene
[2] Toluene/Propan-2-ol (9:1)
[3] Heptanes/Butyl acetate (10:1)
[4] Heptanes/Butyl acetate (3:1)
Retardation factors (RF), from left to right:
[1] ~0.17
[2] 0.69–0.70
[3] A: 0.15; B: 0.19; C: 0.27
[4] A: 0.32; B: 0.40; C: 0.47
[Fig. 131] Samples being dissolved in isopropanol
 [Fig. 132] Developed TLC plates under UV-A
[Fig. 132] Developed TLC plates under UV-A
 [Fig. 133] Developed TLC plates under UV-C
[Fig. 133] Developed TLC plates under UV-C
 [Fig. 134] Recrystallized benzaldehydes (Me / Et \ Pr)
[Fig. 134] Recrystallized benzaldehydes (Me / Et \ Pr)

So, here we are, with all three of the intended benzaldehydes * in modest quantities. I'd be lying if I said I wasn't thrilled with
the result (and having gone through with sharing it to the best of my ability), but I'd also be remiss if I didn't disclose that I am
sweating profusely from the mere thought of still needing to get to my ultimate goal of 3–6 amines with what I have. That said, there are other
targets in the 4-alkylthio realm that I'm interested in (for instance, the rather spooky 2C-T-21), so, for better or for worse, having
another go from scratch isn't entirely unlikely. Or undesired.
* Assuming that PiHKAL contains a couple of bad melting points.
Gulp.
The final 2,4,6-trimethoxybenzaldehyde post will be somewhat more delayed, as I'll be unable to dedicate quite as much of my time to putting it
together.
But it will come.
And it will be nice.
[Edited on 6-1-2023 by Benignium]
|
|
|
SuperOxide
Hazard to Others
Posts: 486
Registered: 24-7-2019
Location: Devils Anus
Member Is Offline
|
|
Bravo, Benignium.
I will read over your latest post in more detail when time permits.
Curious - are you planning on synthesizing every compound in PiHKAL?
[Edited on 5-1-2023 by SuperOxide]
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
Please do, and let me know what you think!
Although I might enjoy trying my hand at synthesizing every single one, biology makes it so that my plan has to involve a degree of cherry-picking,
and not all of the cherries grow on this one tree, either — phenethylamine or otherwise.
|
|
|
pihkameleon
Harmless
Posts: 1
Registered: 20-7-2023
Member Is Offline
Mood: No Mood
|
|
I have previously shared this synthesis on a site which is a little more appropriate for this kind of chemistry, but after some talking with
Benignium, it was decided to polish up the report a litle, add some photographs and post here too. It should serve as a further motivation to get out
and do interesting chemistry (whatever that means to you!), document it, and publish it for the greater good (whatever that means to you!). The
starting compound, 2,5-dimethoxyacetophenone, was a gift from a good friend and was lying around for over a year (during which it turned from slightly
yellow to orange). But it would be easily enough made under the appropriate Friedel-Crafts conditions (a little more gentle than Shulgin, to avoid
demethylation - check [1], the book should be available in its English version on appropriate "shadow libraries"). Had I known how simple it was to
make my target compound in the end, I wouldn't have waited for so long. So, get out and do chemistry now! And don't forget to write home about it!
Please note that this chemistry is completely unoptimised, as often times, I'm more intrigued by the structures of the finished products than the road
towards them. One day, I may fall in love with one of these compounds and decide to really optimise a route - for now I'm ever crushing on the next
hot compound. I am planning to make the benzaldehyde again, and a couple of other compounds derived from it. Maybe this will be a chance for more
meticulous chemistry and better pictures. Concerning the TLC, either dichloromethane or 1,2-dichloroethane was used for elution of the ethylbenzene,
benzaldehyde and nitroalkene. Sometimes, the line for the solvent front was forgotten. I wouldn't trust any of the values you see for Rf anyways, as
all my plates are old and wet. But it should give an idea.
| Quote: | 2,5-dimethoxyethylbenzene [1]
In a 100 mL threeneck-RBF with stirbar, thermometer and distillation head, 10.618 g slightly impure 2,5-dimethoxyacetophenone(1) (MW
180.20, 58.9 mmol) were dissolved in 53 mL ethylene glycol, followed by 7.33 g ground up KOH (MW 56.11, 130.6 mmol, 2.2 eq., probably slightly wet),
followed by 8 mL of hydrazine hydrate (MW 50.06, 164.1 mmol, 2.8 eq.). The flask was immersed in an oil bath and over the course of 3 h, water was
continuously distilled out. When the internal temperature reached 180 °C, the distillation head was replaced by a Liebig condenser, and the contents
refluxed at 180 °C for a further 3 h. Attempts at monitoring the reaction on TLC directly were inconclusive (micro-workup with extraction might be
better!). The heating from the oil bath was discontinued, and the chemist left the reaction to sleep 4 h and work up in the morning. The aqueous
distillate (which contains steamed-over product) was filled up to 100 mL with H2O and combined with the reaction mixture. This aqueous solution was
extracted with 5 x 10 mL CH2Cl2, the combined CH2Cl2 layers dried over 3 A molecular sieves, filtered, solvents removed on the rotovap, and the
remaining yellow-orange liquid vacuum-distilled, collecting the distillate at 86 °C @ ? mbar(2) (lit. 104 - 105 °C @ 7 mm/Hg [1]) as a
slightly yellow liquid. Product was homogenous on TLC and weighed 7.613 g (MW 166.22, 45.8 mmol, 77 % yield).
Remarks:
(1) slight spot on TLC hovering over the acetophenone
(2) one day, I'll figure out how to read mercury manometers

2,5-dimethoxyacetophenone under ethylene glycol

Wolff-Kishner during reflux period - no foaming in sight, gentle

TLC of crude 2,5-dimethoxyethylbenzene from solvent extraction after reaction

crude 2,5-dimethoxyethylbenzene

simple vacuum distillation of 2,5-dimethoxyethylbenzene - lacked equipment (cow adapter/spider) for proper set-up, but there was no distillate before
the product

remaining tar of vacuum distillation

TLC of 2,5-dimethoxyethylbenzene after distillation
|
| Quote: | 4-ethyl-2,5-dimethoxybenzaldehyde [2, 3]
In a 100 mL threeneck-RBF fitted with reflux condenser and addition funnel(1), 2.543 g hexamine (MW 140.19, 18.14 mmol, 2 eq.) and 1.518 g
2,5-dimethoxyethylbenzene (MW 166.22, 9.13 mmol) were dissolved in 12 g AcOH. The flask was put on a cold water bath, and via addition funnel, a
solution of 3.700 g H2SO4 (MW 98.08, 96%, 36.22 mmol, 4 eq.) in 10 g AcOH was dropped in over a timeframe of 20 min. This lead to the precipitation of
a white precipitate. The suspension was put in a hot oil bath and refluxed for 2.5 h (upon heating, the precipitate dissolved and the solution turned
from faintly yellow to yellow to orange), then the oil bath removed, and when the temperature dropped below 100 °C, 25 mL of H2O, followed by 25 mL
EtOAc were added. The solution was stirred overnight (~ 14 h) at room temperature. 5 mL of aq. sat. NaCl were added, phases separated, and the aq.
phase extracted with 4 x 15 mL EtOAc. The combined organic phases were washed with 3 x 45 mL 10 % NaCl solution, then 45 mL dilute K2CO3
(caution!!(2)), then 45 mL brine. The organic phase was dried over 3 A molecular sieves, the sieves filtered off, and evaporated to leave
1.666 g of a yellow oil (MW 194.23, < 8.58 mmol, < 94 %, impure by TLC, contaminations on baseline, eluent 1,2-dichloroethane).
Remarks:
(1) it was started with the addition funnel on the sideneck, but this lead to nasty precipitation at the corner, and as such the reflux
condenser was removed and the addition funnel clamped to drop the solution in directly from above
(2) this step was performed in the seperatory funnel by me. Huge mistake, as there is still appreciable amounts of AcOH dissolved in the
organic phase even with the aqueous washings. A small amount of extract was lost due to pressure build up with closed cork. Probably best to add K2CO3
carefully with stirring in an open vessel until CO2 evolution ceases.

at the relatively start of the addition of H2SO4 - addition funnel was clamped from above, as otherwise precipitate would have formed on the sides and
wouldn't have been stirred, potential of clogging too

finished addition of H2SO4/AcOH, everything precipitated

precipitate gets dissolved during heating, reaction mixture turns from faintly yellow to bright yellow to orange

TLC of reaction mixture during hydrolysis

TLC of extracted benzaldehyde, used as is in the next step - long-wave UV vs. short-wave UV - 4-ethyl-2,5-dimethoxybenzaldehyde shows bright
fluorescence
|
| Quote: |
4-ethyl-2,5-dimethoxynitrostyrene (4-Et-2,5-DMNS) [4]
Above oil was used without further purification(1) (calculations for equivalents assume 100 % purity though!). The oil was dissolved in a
mixture of 1.101 mg ethanolamine (MW 61.08, 18.03 mmol, 2.1 eq.) and 2.150 g AcOH (MW 60.05, 35.80 mmol, 4.17 eq.), diluted with 2.5 g propan-2-ol.
0.618 g nitromethane (MW 61.04, 10.12 mmol, 1.18 eq.) were added, and the mixture stirred at r.t. for 3.5 h, after which TLC showed completion (check
via long-wave UV, where the benzaldehyde shows bright fluorescence!). The thick suspension with orange-yellow precipitate was diluted with 4 volumes
of H2O, stirred for 10 min, and then filtered and washed with copious amounts of H2O. The resulting orange-yellow precipitate was dried crudely on the
pump, and then recrystallised from excess isopropanol (~20 mL) for a week at -18 °C(2). After filtration and evaporation of solvents on
the rotovap, this resulted in 1.125 g of 4-ethyl-2,5-dimethoxynitrostyrene (MW 237.26, 4.74 mmol, 55 % yield over two steps).
Remarks:
(1) the impure benzaldehyde was used because I trust these kind of conditions for clean nitroalkene synthesis, and because I was impatient.
Would not recommend, given that bisulfite adduct will easily lead to a good product.
(2) random amount until I had time for the laboratory again

precipitated nitrostyrene after 3.5 h

TLC of reaction progress - compare fluorescent benzaldehyde to nitroalkene
|
| Quote: |
4-ethyl-2,5-dimethoxyphenethylamine hydrochloride (2C-E HCl) [5]
In a 100 mL Erlenmeyer flask with efficient stirbar and Liebig condenser in a cold water bath, 1.335 g NaBH4 (MW 38.83, 34.38 mmol, 7.25 eq.) are
dissolved in a mixture of 23 mL isopropanol/H2O 2:1. Over a timeframe of 10 min, 1.125 g 4-Et-2,5-DMNS (MW 237.26, 4.74 mmol) are added in small
portions to the reaction mixture (by temporarily disconnecting the reflux condenser). After the addition, the water bath is removed, and the condenser
re-attached. After 15 min of stirring at r.t., the flask is immersed in an oil bath, and heated up to reflux. When the oil bath reached 60 °C, 140 mg
Cu(OAc)2 * H2O (MW 199.65, 0.70 mmol, 0.15 eq.) dissolved in 3 mL H2O are introduced rapidly by temporarily disconnecting the reflux condenser. The
reaction is brought to reflux and stirred at reflux for 50 min. After cooling down to r.t., 3 g NaOH in 10 mL water are added. The organic and aq.
phase are separated(1), and the aq. phase extracted with 2 x 15 mL isopropanol. The combined organic phases are washed with 3 x 20 mL aq.
sat. K2CO3, then separated and slightly acidified by dropwise addition of conc. aq. HCl. The solvents are removed on the rotovap, this is aided by 2x
addition of 10 mL IPA shortly before dryness. The resulting yellow salts are dissolved in 60 mL H2O, washed with 3 x 20 mL EtOAc (an emulsion broken
by addition of a bit of sat. NaCl). The aqueous phase is basified with 3 g NaOH in 10 mL H2O, then extracted with 3 x 20 mL EtOAc, and the EtOAc
washed once with 20 mL NaCl brine. The combined organic phases are dried over 3A molecular sieves, the sieves filtered off, then the solvents
evaporated, yielding a not perfectly free of solvents slightly yellow oil weighing 918 mg. This is dissolved in ~15 mL isopropanol, neutralised with
conc. aq. HCl(2), then 3 volumes acetone are added and the suspension dumped in the freezer for way too short time (1 h)(3).
After filtering, washing with acetone and drying on the rotovap, this results in 691 mg 2C-E HCl (MW 245.7, 2.81 mmol, 59 % yield, mp 211 - 212 °C)
as fine powdery white crystals.
Remarks:
(1) it is better practise to first filter off the cuprous sludge, but in some cases it can be omitted... not recommended though!
(2) even before addition of acetone, this turned into an almost solid chunk of crystals
(3) the pihkameleon has many time restraints for his laboratory playtime!

~ 100 mg of 4-ethyl-2,5-dimethoxyphenethylamine hydrochloride on a scale
|
Literature
[1] Trachsel, D. Psychedelische Chemie: Aspekte psychoaktiver Moleküle, 5. Auflage.; 2016. ISBN: 978-3-907080-53-5
[2] https://www.thevespiary.org/talk/index.php?topic=2840.msg286...
[3] https://www.sciencemadness.org/talk/viewthread.php?tid=15749...
[4] https://www.thevespiary.org/talk/index.php?topic=17911.0
[5] https://www.thevespiary.org/talk/index.php?topic=15090.0
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
The pictures and overall layout do a fantastic job at complementing the impressive experimental work that you've done, pihkameleon! Thank you for
posting this!
|
|
|
Benignium
Hazard to Others
Posts: 115
Registered: 12-6-2020
Member Is Offline
Mood: Quasi-catatonic
|
|
For your consideration:
2,4,6-trimethoxybenzaldehyde
196.20 g/mol
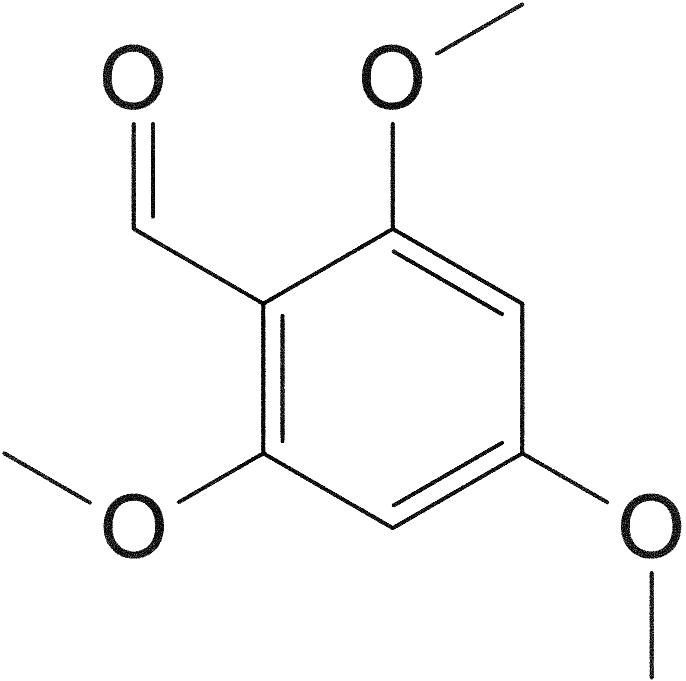
In 1884, researchers Wilhelm Will and Karl Albrecht of the Friedrich Wilhelm University in Berlin published an
article [1] in the scientific journal Berichte der Deutschen Chemischen Gesellschaft
regarding their ongoing work on assigning the appropriate trihydroxybenzene building blocks to two natural dioxycoumarin compounds, esculetin and
daphnetin. So far, daphnetin was known with certainty to be derived from pyrogallol (1,2,3-benzenetriol), but whether its constitutional isomer
esculetin conformed to the structure of hydroxyquinol (1,2,4) or phloroglucinol (1,3,5) was still up for debate. The scope of their publication,
therefore, was to identify the specific benzenetriol component of esculetin, while simultaneously producing precious experimental data on the
derivatives through which they intended to do so. In essence, three distinct triethoxybenzoic acid isomers and their ethyl esters were to be formed
from pyrogallol and phloroglucinol, [A] which could then be compared with analogous derivatives of
the unidentified benzenetriol obtained from natural esculetin. [B]
[Fig. 1] Structures of daphnetin and esculetin
In a surprising turn of events, when the carboxylic acid derivative of phloroglucinol was dissolved in ethanol and subjected to a
strong acid catalyst in an attempt to form the ethyl ester, [C] an evident decarboxylation took
place instead. Moreover, the observed liberation of gaseous carbon dioxide was accompanied by a further transformation into something which was found
to crystallize as long, white needles. This new material withstood heating in alcoholic alkali, and its reaction with ferric chloride was neither the
intense blue of the phloroglucinic acid nor the violet that was expected of phloroglucinol. Fascinatingly, it turned out that the main product of the
procedure was, in fact, the diethyl ether of phloroglucinol. [D] What's more, it was also
recognized that unlike phloroglucinol, this diethylated material could be exhaustively ethylated via the conventional Williamson ether synthesis to
obtain pure 1,3,5-triethoxybenzene, whose novel melting point would then inform the accurate conclusion that esculetin bore the structural motif of
hydroxyquinol. This pioneering work with its serendipitous discoveries garnered significant attention toward the onionesque morass that was
phloroglucinol; an issue that would vex chemists for decades to come; enduring so far as to stare back at me from the pages of PiHKAL [2] — and while the thick, proverbial jungle which once dictated these paths of quasi-alchemical
adventure and niche camaraderie may be long-macadamized by the migratory march of science, the occasional unsuspecting amateur remains likely to have
their jimmies rustled by the perpetually gaping manhole of yesteryear.
But indeed, what does it mean?
In the context of this post, it is crucial to know that attempts at electrophilic methylation of the deprotonated phloroglucinol are
prone to ending miserably: instead of the desired trimethyl ether, such a reaction would afford an overwhelming number of side products, with the
neutral portion consisting mostly of alicyclic materials like 2,2,4,4,6,-pentamethylcyclohexane-1,3,5-trione, due to rampant nuclear alkylation at the
three α-carbons. While the relative quantities of the formed side products are wildly unpredictable, the hexamethyltrione has been demonstrated as
the major (ultimate) product. By all accounts, isolating any fraction of formed 1,3,5-trimethoxybenzene from this mixture would be deemed as an
exercise in futility. [3] [4] [5] [6]
The issue was quickly likened to a ketone-like behavior by Adolf von Baeyer, who, in 1886, described the formation of the
trioxime of phloroglucinol, and proposed that the facultative intermediary be called pseudophloroglucinol; [7] the term tautomerism had just been coined by Conrad Laar, in 1885, but was initially met with a pseudo
label of its own. [8] By forming the trioxime, Baeyer had experimentally proven the existence of
cyclohexane-1,3,5-trione — the triketone tautomer of phloroglucinol — whose exact identity and implications nevertheless remained indeterminate.
At the time, experimental chemistry was in many ways constrained by its severely nascent theoretical foundation. Thus, finding new and improved ways
through this particularly loosely comprehended polyolic black box of tautomerism would continue to rely on tentative experimentation for
decades to come.
In the meantime, the early 20th century arrived, bringing with it infrastructural breakthroughs like the mass production of
affordable cars, widespread electrification, electric toasters, and sliced bread; all of which contributed to an unprecedented rate of scientific
development. In terms of chemistry, on the other hand, a revolution blossomed within the confines of the mind: Spurred, in part, by the maturation of
August Kekulé's autophagous snake anecdote into the driving force behind Johannes Thiele's rudimentary yet pioneering prediction of resonance in
1899, [9] [10] those who sought chemical understanding at the atomic level would draw it
increasingly from theoretical physics, and in doing so would align the two previously segregated fields toward an increasingly common goal. This shift
in philosophical paradigm facilitated the formulation of groundbreaking heuristic techniques like the Lewis structure, and built the foundations of
quantum chemistry. Then, finally—through the concordant invention of modern experimental tools like the concept of pH and NMR spectroscopy, that
could supersede traditional staples like elemental analysis and spruce chips wetted with acid and alcohol [11]—the stage was set for demystifying phloroglucinol once and for all.
In 1956, by comparing the ultraviolet absorption spectra of increasingly alkaline phloroglucinol solutions with those obtained
previously for 1,3,5-benzenetriamine at various stages of protonation, Hans Köhler and Günther Scheibe discovered correlations which led them to
deduce the existence of analogous isosbestic points, and, notably, to predict the large prevalence of a ketonic dianion form through comparison with
the dianion of filicinic acid, whose structure was known. [12] Corroborating the same phenomena,
studies making use of 1H NMR in the 60s would then independently reveal ring protonation to form σ-complexes (i.e.,
arenium ions) in both superacidic [13] and alkaline [14] media. More recently, in a 1993 report by Martin Lohrie and Wilhelm Knoche, UV-visible and diode-array
spectrophotometers were augmented with stopped-flow apparatus and temperature jump techniques in order to observe the chemical kinetics and
thermodynamics of altering the pH in aqueous solutions of phloroglucinol; thereby arriving at a wonderfully comprehensive view of the different
equilibria pertaining to its dissociation and tautomerism (fig. 2). [15]
[Fig. 2] Illustrated species of phloroglucinol
The above Scheme II illustrates most of the structurally distinct species of phloroglucinol. Each
horizontal row depicts the different degrees of dissociation (i.e., of protonation/deprotonation) within a single degree of conjugation; with
each vertical column therefore representing the range of conjugation within a single degree of dissociation; and with structures A
and B being intermediates for acid-catalyzed reactions of 1 to 7 and 7 to
9. On adjusting the pH in either direction, two distinct kinds of reaction were observed to take place: the horizontal double arrows
indicate diffusion-controlled (unmeasurably fast) acid–base interactions that occur independently of tautomerism, whereas diagonal arrows denote the
associated rate-determining keto–enol rearrangements. The embedded Figure 9 presents the species' relative concentrations
on a logarithmic scale as functions of the pH.
In the following paragraphs, I've attempted to explain the phloroglucinol issue in a way that might have enabled myself to more
rapidly grasp the basic concept some ten months ago; for this, I'll continue referring to structures from the above illustration as corresponding
digits in bold.
From the perspective of organic synthesis — specifically, of electrophilic O-alkylation in polar, protic media — in order for an
electrophile (like the dimethyl sulfate whose application is described in this post) to get attacked by the nucleophilic oxygens of phloroglucinol,
each phenol needs to be activated through basic deprotonation; much like has been done to the previous phenols of this thread. This removal
of what is essentially the bare nucleus of a hydrogen (and a major building block in heavier nuclei) results in the negatively charged phenolate
(i.e. phenoxide) anion—the conjugate base of phenol (i.e., a better nucleophile than phenol)—enriched by a third pair of
unbonded valence electrons, which, albeit delocalized due to being conjugated with the aromatic system (i.e., occupying a p-orbital on an
oxygen that is coplanar and in phase with an adjacent benzene carbon), remains subject to the inductive attraction which oxygen exhibits owing to the
higher number of the positively charged protons contained in its nucleus compared to the larger carbon (i.e., its higher electronegativity).
This added charge density seems particularly significant in the phloroglucinol structure, as the three ortho- and para- (but not
meta-)activating phenols are all located meta to each other, leaving the three α-carbons in their midst unusually electron-rich
through π donation such that C-alkylation has been shown to occur and even predominate in the absence of base; when the neutral molecule exists
almost exclusively in its aromatic form, 1. [16] The phenolate monoanion
2 (and by extension the appropriate monoether anion), in contrast, has been successfully reacted with dimethyl sulfate in the
presence of carefully controlled quantities of alkali to produce the O-permethylated ether. [3] [17] [E]
As far as the dianion is concerned, however, it is by now evident that the prediction of a prevailing dibasic phenolate form
(3) is turned on its head by the phenomenon referred to as keto–enol tautomerism—the proton-transfer equilibrium enabling
transformations between a nucleophilic enolic form (an alkene situated adjacent to an alcohol) and an electrophilic
ketonic form (a carbonyl with an accommodating α-carbon), catalyzed by both donors and acceptors of protons (i.e., acids and bases in the
Brønsted sense, respectively). Judging by the previous observations of predominant C-alkylation, it follows, therefore, that the prevalence of the
ketone-like character following a second deprotonation is high enough to almost completely exclude the oxygen atoms from interacting with the
alkylating agent — while the α-carbons move up in pecking order, collectively consuming (in total) up to six molar equivalents of the
same.
At a glance, the aromaticity that is ubiquitous in phenols might be expected to favor a desirable polyenolic arrangement — after
all, the equivalent of this is known to be the case even for the symmetric benzenediol, hydroquinone, whose stability is accordingly greatest when
there is a continuous ring of conjugated p-orbitals in a planar conformation (i.e., aromaticity) — However, rather than going from
1 → 2 → 3 → 4 in a straightforward series of deprotonations, as the already unusual electron density of phloroglucinol is made
even greater, it begins rearranging in exquisite ways to compensate, promptly doing away with its aromatic character; the urgency with which these
rearrangements take place is equally striking, and likewise reflected by the overlap between the first two of its acid dissociation constants,
pKa1 = 8.6–9.0 and pKa2 = 8.9–9.1 (with pKa3 = 14). [15] Nevertheless, even when staring at the above Scheme II, the reasoning behind this observed
inclination is not immediately obvious.
In mulling the subject over, guidance was sought, and two hints were received which were found specially fruitful.
The first one points to structures with two carbonyls sharing an α-carbon: A well-established quirk of nature, these are commonly
referred to as 1,3-dicarbonyls or (where applicable) β-diketones, and are almost universally introduced at a point during university lectures on
organic chemistry in the form of a β-keto ester product of a Claisen condensation. Belonging under a larger phenomenological classification called
active methylene compounds [where there’s a methylene in between two electron-withdrawing groups (e.g. -NO2, -CN, -COOR)], their
distinctive feature is an sp3-hybridized carbon center with particularly labile (i.e. acidic) hydrogens, whose dissociation gives
a highly resonance-stabilized conjugate base; communicated via a general approximation of pKa = 9. Indeed, implicit postulates of such
a diketonic anion are many for phloroglucinol (as well as resorcinol and other similarly related substances), and the structures of Scheme
II approach it as closely as limiting the portrayals of negative charge to the oxygens seems to allow.
The other concept deserving of a good grasp has to do with the stability of a structure as it relates to bond (dissociation)
energies. On its own, this value, determined for a given bond at its equilibrium length, measures the amount of energy that would be required to
completely negate the interactive forces between two covalently bonded atoms by pulling them apart. Consequently, the sum of these individual values
contained within a given molecule can be calculated to evaluate the stability and thermodynamic favorability of said molecule. The total energies were
calculated for the kinetic dianion (3) and the proposed thermodynamic dianion structure according to values obtained from a single
literature source. [18] As such, these came out to be 5590–5804 kJ/mol and 5738–5959 kJ/mol,
respectively, which would tip the scales in favor of the diketone tautomer. However, after correcting the former value through incrementing it by 151
kJ/mol according to the aromatic stabilization observed for benzene, the two values appear to converge, obfuscating whatever evidence is
there.
It is my belief that the combined bond energy of the proposed thermodynamic product is complemented by a superior distribution of
charge between a single enolate anion and the 1,3-dicarbonyl anion whose smear of electron probability (another cool way to think about
electron density) quite exactly opposes the more localized charge of the former, reducing the overall dipole moment of the structure. Below is a
somewhat streamlined illustration of the logistics for arriving at the discussed dianion structure as I currently understand them; referring to
Scheme II, this progression would correspond to 1 → 2 → 3 → 6, at which point it splits into two
distinct routes via 5, either through 8 or by way of resonance. Alkylation (particularly involving the smaller, less
sterically hindering alkyl groups) of the dissociated active methylene in between the carbonylic oxygens will serve to further stabilize the charge
toward the substituted α-carbon through σ donation by the alkyl substituent—facilitating geminal alkylation prior to a structural or
canonical (i.e., mesomeric, as in "resonance contributor") shift to favor another one of the ring carbons in the same way. The
six-membered ring structure of the dianion favors alkylation at the carbon by constraining the 1,3-dicarbonyl anion into a W-shaped conformation,
where the nucleophilic middle protrudes, and the carbonylic oxygens are pointed away such that they do not hinder its access or affinity toward
electrophilic substrates; moreover, this conformation minimizes the dipole-dipole repulsion at the site, which contributes to stability.
[Fig. 3] Favorable pathways to the thermodynamic dianion structure
But wait, there’s more!
What has been discussed on the theory so far leaves out a bunch of nuance which I’d like to touch on briefly before a few more
words on the practical side of things.
As previously stated, the above focuses on changes that take place in prot(on)ic (i.e. hydrogen-bonding) solvents like water
and alcohol. These can interact with the anionic species of phloroglucinol and its alkylated derivatives, as well as the cation and the alkylating
agent. Interaction like this can significantly favor C-alkylation by solvating (i.e. surrounding) and thus effectively shielding the oxygen
of an enolate anion; similarly, it can also hinder the desired reactivity by encumbering the alkylating agent. Additionally, a protic solvent can act
as an acid or a base in tautomeric proton-shifts. The use of polar solvents in this context is a given, since a heterogenous reaction with an
undissolved solid salt (whose oxygen anion is shielded by the associated cation in the crystal lattice) would vastly favor C-alkylation. The use of
aprotic solvents—and especially dipolar aprotic ones [i.e., ones with a sizable dipole moment (e.g. DMF, DMSO,
MeCN)]—on the other hand, tends to favor O-alkylation through not only the lack of hydrogen-bonding, but also a proportionally stronger solvation of
the metal cation relative to the enolate anion, which is consequently freer to attack with its more electronegative atom. [18] [19]
Furthermore, the concept of hard–soft character (i.e. polarizability) in Lewis acids and bases has been found to correlate
with a superior selectivity for O-alkylation exhibited (in some solvents) by dialkyl sulfates, whose hard sulfate leaving group is more amenable to
coordinating (i.e. bonding) to the hard oxygen donor atom of an ambivalent metal enolate in comparison to a softer vicinal carbon donor.
Conversely, the soft character of alkyl halides (which decreases with the atomic radius of the halogen) explains why their involvement favors
C-alkylation. It is also worth noting that the carbon under nucleophilic attack in an SN2 transition state favors attack by the less
electronegative site of an ambident nucleophile. Among the hard Lewis acids that are most often associated with a phenolate anion, the size of the ion
has a significant impact on selectivity, whereby increased bulk (e.g., K+ > Na+ > Li+) corresponds to
an increased tendency to dissociate, and therefore a higher reactivity of the anion as a nucleophile. [18] [19]
In the gas phase, where the enolate anion (of cyclohexanone) is completely free, there is only O-alkylation. [18]
A striking irony of the phloroglucinol subject is that so much of this newfound understanding on it from the past century seems to
have had no bearing on the seldom undertaken laboratory preparation of its trialkyl ethers. The most commonly encountered procedure (among the amateur
community, at least) still seems to be the same one that is featured in PiHKAL (which itself is only a slight variation of the 1884 procedure by Will
and Albrecht), where phloroglucinol is reacted with an excess of alcohol in the presence of a strong acid catalyst; [F] followed by a bimolecular nucleophilic displacement (SN2) reaction of the resulting dialkoxyphenol with an
electrophilic alkylating agent in the presence of a base (i.e., the Williamson ether synthesis). Nevertheless, interesting experimental
discoveries have been made. In closing, here are some of the highlights:
As early as 1900, F. Kaufler suggested that the deprotonated phloroglucinol acts as a ketone toward methylation, but as a phenol
toward benzylation; [20] a seemingly reasonable observation of steric effects, whose preciseness
has since been disputed. [21]
In 1920, K. Freudenberg found that reacting the phloroglucinol diethyl ether with 5.32 molar equivalents of dimethyl sulfate (based
on phloroglucinol) gave improved yields of up to 80%. [22] [G]
A 1953 publication by H. Bredereck, I. Hennig and W. Rau reports success in circumventing the C-alkylation-favoring tautomeric forms
by employing a dropwise addition of 20% NaOH to a mixture containing 3.86 molar equivalents of dimethyl sulfate and 1.9 mL of acetone per gram of the
anhydrous phloroglucinol while maintaining a constant pH in the range of 8–9; colorless crystals corresponding to a 93% yield were reported [17] [H] — in contrast, a dropwise addition of alcoholic base had previously been trialed by J. Herzig
and F. Wenzel in 1906, with modest yields and some C-alkylation being reported. [3]
Arguably the most convenient-sounding procedure for direct trialkylation was presented by J. W. Clark-Lewis in 1957, involving the
portionwise addition of DMS to a mixture of anhydrous phloroglucinol and potassium carbonate in acetone. [23]
The esterification using acetic anhydride proceeds in a straightforward manner to yield the 1,3,5-triacetoxybenzene, which has been
subjected to alkylation in the presence of base so that the ethers form just as the electron-withdrawing acetates are saponified, keeping the electron
density low enough to avoid C-alkylation. [6]
Finally, I would like to state my heartfelt gratitude for my fellow SM member AvBaeyer, whose
compassionate gift of awe-inspiring expertise was pivotal for my learning and composing of the above.
Notes
[A]: The carboxylic acid moiety was introduced, in yields that were reported as nearly quantitative, by digesting the
benzenetriols [in water, at 130°C, in a pressure reactor] with four equivalents of ammonium carbonate (or in the case of phloroglucinol, with
potassium carbonate).
[B]: In a later publication, Will outlines the processing of naturally derived glycosidic materials, including
esculetin, as follows: A water-soluble cleavage product, usually a phenolic ester, is first saponified using potassium hydroxide, followed by
treatment with methyl (or ethyl) iodide at reflux. The resultant ether is oxidized with potassium permanganate to remove the unwanted, mostly
unsaturated aliphatic ring substituents. A dry distillation of the calcium carboxylate derivative is then employed to isolate the corresponding phenol
ether, whose constitution (once further purified via steam distillation, in the case of esculetin) can be determined by comparison with the
synthetically prepared phenol ethers. Additionally, this article reports the purification of 1,3,5-trimethoxybenzene via steam distillation after
adding some KOH solution. [24]
[C]: Interestingly, although the process is very similar to the Fischer–Speier esterification described in 1895 (also
in Berlin), it is credited to Hugo Schiff, who in turn refers to the preparation of ethyl gallate by the French chemist Édouard Grimaux in
1864.
[D]: More specifically, a mixture of products was determined which contained some 10% of the monoether. [25]
[E]: Since the monoanion of phloroglucinol has been shown to not predominate at any pH, [15] I suspect that the successes of these procedures may be largely due to its diffusion-controlled formation and subsequent
ability to react immediately in the presence of excess electrophile.
[F]: The formation of phloroglucinol mono- and dialkyl ethers in Fischer–Speier-like conditions is thought to occur
through a ketal intermediate: [26] a keto tautomer (initially 7 from
Scheme II) first undergoes the addition of an alcohol across a protonated carbonyl, followed by a deprotonation to give a
hemiacetal; a protonation of the hydroxyl then facilitates its elimination as water, allowing for a renewed C–O π bond, a repeated protonation of
the oxygen, the addition of a second alcohol, and the formation of the ketal through a final deprotonation. However, since a neutral ether can result
via elimination from either an acetal or a hemiacetal, the latter would be more likely owing to the fewer steps involved. Interestingly, the steps of
an acetalization have been shown to occur in a concerted fashion. [27]
[G]: The high consumption of methylating agent is probably due to the presence of the monoether, which is still very
much susceptible to C-alkylation.
[H]: This is a likely origin for the claim, presented by GC_MS on The Hive [cited in the permethylation chapter (III)
of my previous post], where a protective effect against the saponification of dimethyl sulfate by aqueous alkali was ascribed to the presence of 2
mL/g of acetone. Here, however, the authors merely suggest a possibility.
References
[1]: W. Will, K. Abrecht (1884): Ueber einige Pyrogallussäure- und Phloroglucinderivate und die Beziehungen derselben zu
Daphnetin und Aesculetin.
https://doi.org/10.1002/cber.188401702110
[2]: A. Shulgin, A. Shulgin (1991): PiHKAL. (#162 TMA-6)
https://isomerdesign.com/PiHKAL/read.php?id=162
[3]: J. Herzig, F. Wenzel (1906): Studien über Kernalkylierung bei Phenolen.
https://doi.org/10.1007/BF01525129
[4]: A.R. Stein (1964): THE METHYLATION OF THE RESORCINOLATE DIANION.
https://doi.org/10.1139/v65-200
[5]: J. Herzig, B. Erthal (1910): Notiz über die Darstellung des Hexa- und Pentamethylphloroglucins.
https://doi.org/10.1007/BF01530256
[6]: H. Kawamoto, F. Nakatsubo, K. Murakami (1996): O-BENZYLATION OF PHLOROGLUCINOL VIA PHLOROGLUCINOL TRIACETATE.
https://doi.org/10.1080/00397919608003645
[7]: A. Baeyer (1886): Ueber das Trioxim des Phloroglucins.
https://doi.org/10.1002/CBER.18860190145
[8]: J.W. Baker (1934): Tautomerism.
https://doi.org/10.1038/135247a0
[9]: J. Thiele (1899): Zur Kenntniss der ungesättigten Verbindungen. Theorie der ungesättigten und aromatischen
Verbindungen.
https://doi.org/10.1002/jlac.18993060107
[10]: F.E. Ray (1934): Thiele's Theory of Partial Valency in Terms of Electrons.
https://scholarworks.uni.edu/pias/vol41/iss1/43/
[11]: A. Bayer (1885): Ueber die Synthese des Acetessigäthers und des Phloroglucins.
https://doi.org/10.1007/BF01518276
[12]: H. Köhler, G. Scheibe (1956): Übergänge zwischen aromatischen und aliphatischen Formen bei Benzolderivaten.
https://doi.org/10.1002/zaac.19562850315
[13]: A.J. Kresge, G.W. Barry, K.R. Charles, Y. Chiang (1962): The Protonation of Phloroglucinol and its Ethers: An Exception to
the Acidity Function Concept.
https://doi.org/10.1021/ja00881a028
[14]: R.J. Highet, T.J. Batterham (1964): The structure of the phloroglucinol dianion.
https://doi.org/10.1021/jo01025a501
[15]: M. Lohrie, W. Knoche (1992): Dissociation and Keto-Enol Tautomerism of Phloroglucinol and Its Anions in Aqueous
Solution.
https://doi.org/10.1021/ja00056a016
[16]: A. Gissot, A. Wagner, C. Mioskowski (2004): Buffer-induced, selective mono-C-alkylation of phloroglucinol: application to
the synthesis of an advanced intermediate of catechin.
https://doi.org/10.1016/j.tet.2004.06.026
[17]: H. Bredereck, I. Hennig and W. Rau (1953): Alkylierungen mehrwertiger Phenole mit Dialkylsulfat.
https://doi.org/10.1002/cber.19530860909
[18]: M.B. Smith, J. March (2007): March’s Advanced Organic Chemistry.
(6th edition; p. 29, 513–519)
[19]: H.O. House (1972): Modern Synthetic Reactions.
(2nd edition; p. 509–529)
[20]: F. Kaufler (1900): Über den Einfluss der eintretenden Radicale auf die Tautomerie des Phloroglucins.
https://doi.org/10.1007/BF01526082
[21]: E. Deme (1976): Synthesis of Authentic Tri-O-benzylphloroglucinol.
https://doi.org/10.1021/jo00885a031
[22]: K. Freudenberg (1920): Über Phloroglucin Gerbstoffe und Catechine.
https://doi.org/10.1002/cber.19200530818
[23]: J.W. Clark-Lewis (1957): PREPARATION OF PHLOROGLUCINOL TRIMETHYL ETHER.
https://doi.org/10.1071/CH9570505
[24]: W. Will (1888): Ueber einige Reactionen der Trimethyläther der drei Trioxybenzole und über die Constitution.
https://babel.hathitrust.org/cgi/pt?id=hvd.cl1hzt&seq=65...
[25]: J. Pollak (1897): Einiges über die Äther des Phloroglucins und eine Synthese des Hydrocotoins.
https://doi.org/10.1007/BF01518276
[26]: J.C. Touchstone, J. Ashmore, M.N. Huffman (1956): Ethers of Phloroglucinol.
https://doi.org/10.1021/ja01602a049
[27]: E. Grunwald (1985): Reaction mechanism from structure-energy relations. 2. Acid-catalyzed addition of alcohols to
formaldehyde.
https://doi.org/10.1021/ja00302a019
[a4] 3,5-dimethoxyphenol
Also called: Phloroglucinol dimethyl ether
154.16 g/mol
Like so many others, I also opted for the procedure outlined in PiHKAL. [1] At the time of my
experimentation (May, 2022), it never occurred to me to attempt to purify this product, and the fact that it would form crystals became clear only
when composing this post. Removal [or enhanced conversion (via completely trapping/removing all water past the hemiacetal formation, or by repeating
the acetalization procedure)] of the monoether should work to minimize the portion of methylating agent that is wasted on C-alkylation in the next
step; quite possibly improving the yield.
[1]: PiHKAL: #162 TMA-6. https://isomerdesign.com/PiHKAL/read.php?id=162
Experiment 1
Phloroglucinol dihydrate (12.99 g, 80 mmol) was dehydrated to constant weight in a 100°C oven [1] and dissolved in methanol (42 mL) in a 100 mL conical flask. With good magnetic stirring, concentrated
sulfuric acid (7.1 mL, 131 mmol) was added from a dropper, causing the mixture to heat up to 45–50°C and take on a vibrant red coloration. After
first stirring it at room temperature for two hours, the mixture was refluxed for ~8 hours and then allowed to cool.
[1]: Admittedly, this step (on its own) was probably unnecessary; with the excess of alcohol that was used, the reaction doesn't
appear to be that sensitive to moisture.
[Fig. 4] Commercial phloroglucinol dihydrate

[Fig. 5] Reaction mixture following acid addition

Work-up
The cooled reaction mixture was diluted with 65 mL of water. Two extractions were done using 25 mL portions of
dichloromethane. The pleasant-smelling [1] extract was dried over some calcium chloride and
filtered to remove the solids. This was then stored in the dark, at room temperature, for 14 days, after which it was concentrated; first by
distilling it down to half its original volume, and finally by stirring the remainder magnetically on a 45°C hotplate while argon was gently blown
into the flask to carry out the residual solvent. A clear, viscous red oil remained, weighing 7.89 g.
[1]: The odor of the extract was a mixture of floral indole and synthetic wild strawberry essence. There was a definite shared
resemblance to methyl salicylate and (low vapor concentrations of) methyl anthranilate in terms of sweetness and texture.
[b5] 1,3,5-trimethoxybenzene
168.19 g/mol
With the foretold red oil in hand, I thought it the perfect opportunity to revisit the solvent free methylation [1] whose first replication involving the solid methylhydroquinone substrate (a3) was less than representative.
[1]: https://patents.google.com/patent/US4065504A/en
Experiment 1
To the 7.89 grams of red oil from the previous step, there was added potassium carbonate (39.49 g, 286
mmol), [1] followed by dimethyl sulfate (30.1 mL, 318 mmol), and the vessel was equipped with a
water-cooled 200 mm Liebig condenser, which was topped with a bulbous anti-splash adapter. The mixture was stirred magnetically, its temperature
remaining at ~26°C with no external heat being applied; its mobility improved over ~20 minutes as the beads of base grew less clumped together.
Heating was commenced after the first hour to counteract a thickening consistency, bringing the mixture to ~35°C on a 60°C hotplate. The gradual
addition of water was also deemed appropriate at this time, and was begun, five drops at a time. [2] 20 minutes later (at the 90-minute mark), 1.4 g of water had been added. The reaction temperature had reached 41°C, and
the hotplate setting was raised to 80°C. Very suddenly, the rate of the reaction picked up dramatically, and the reaction temperature shot up to
75°C, by which point the heating was discontinued; dense fumes were observed exiting the apparatus, as the peachy gruel in the flask below curdled
into a white, clumpy porridge from which a clear, red liquid phase separated. [3] Larger,
approximately half-gram portions of water were added to just barely keep the stir bar functional. [4] Heating was resumed once the mixture had cooled to 66°C. Two hours into the reaction, a thinner, more homogenous
consistency had been regained; 7 g of water had been added, and the internal temperature was a stable ~60°C on the 100°C setting — and yet, a slow
thickening persisted, which prompted the addition of three more grams of water. Finally, the mixture was kept agitated for a little over 15 more
hours, with the hotplate set to 125°C.
[0]: The temperatures were measured from the exterior surface of the flask using IR.
[1]: This time, the carbonate had been briefly ground down to a slightly finer consistency.
[2]: 10 g of water had been pre-measured for use as a lubricant in near-accordance with the patent that was followed; the
instructions are to use an amount that is "at most equal to, but usually considerably less than, the initial weight of the hydroxybenzene compound".
In hindsight, I do not believe that 7.89 grams would have been sufficient here — but then, I suspect that the embodying reactions of the patent were
stirred mechanically.
[3]: I suspect that this sudden exothermic eruption was mostly due to C-alkylation of the monoether, to which the added water would
seem likely to have contributed. Moreover, the observed generation of chromophores and the persisting reddish oil (during workup) would match
literature descriptions of the C-alkylated pseudoethers.
[4]: Although the stir bar spun very fast and never decoupled, the parts of the mixture that had risen up against the sides of the
flask stagnated momentarily as the reaction hit the peak of its vigor. Barring mechanical stirring, a larger (high-torque) stir bar could avoid this
problem, particularly if powdered K2CO3 was added to a stirred mixture of the liquid components in the beginning (however, the
presence of the monoether bears consideration). One might also be more liberal in their use of the allocated water early on, and consider using a
water bath that can act as a heat sink such that there is more time to react, should the reaction suddenly sustain itself and then some. Having said
all that, a second attempt by me would avoid the hassle and just use acetone as solvent.
[Fig. 6] Addition of potassium carbonate to crude diether

[Fig. 7] 30 minutes after DMS addition

[Fig. 8] 1:15 into the reaction

[Fig. 9] (1:38) Sudden increase in reaction vigor

[Fig. 10] (1:52) Immediate aftermath
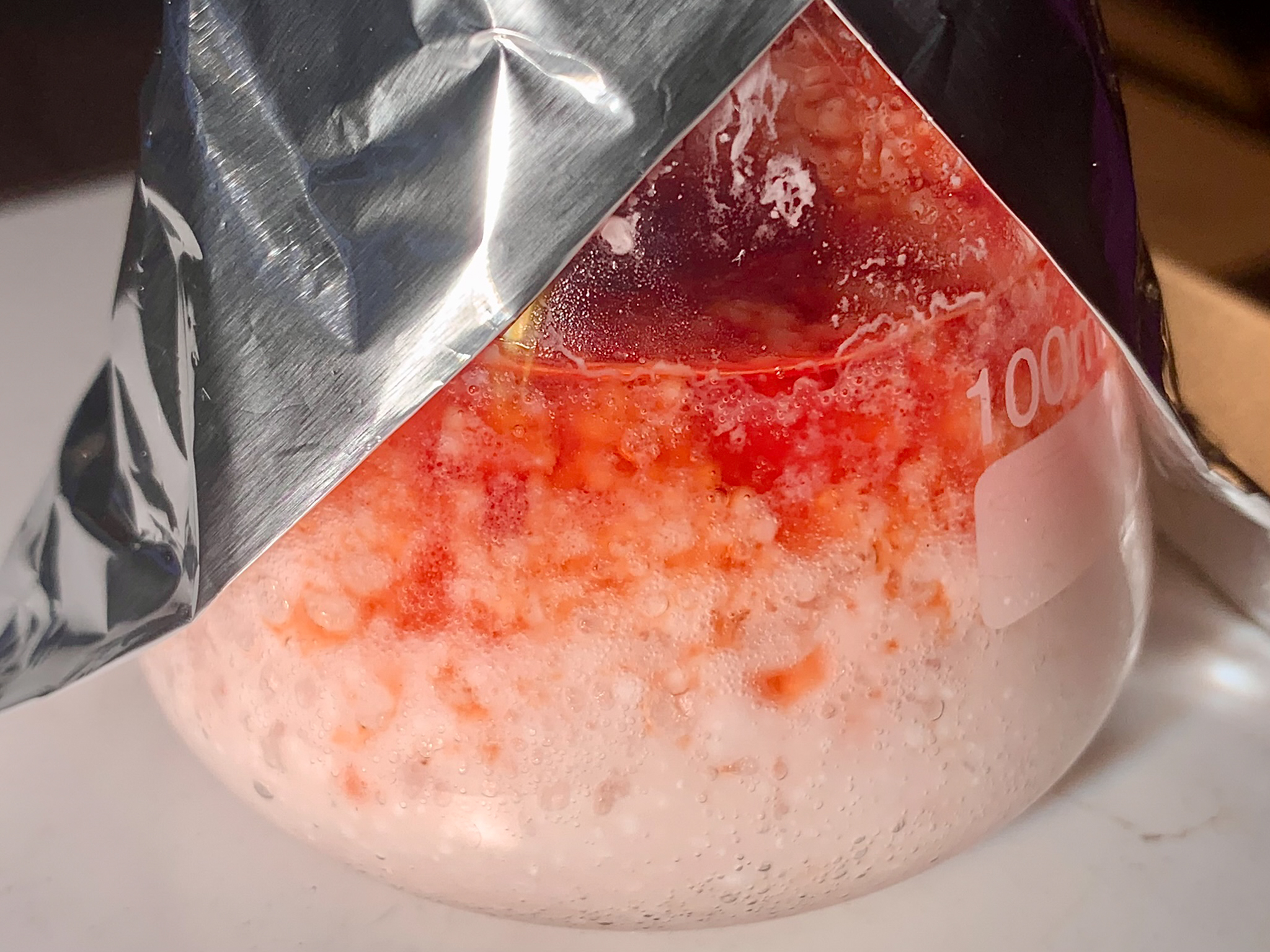
[Fig. 11] Reaction mixture following complete water addition

Work-up
On the following day, 50 mL of water was used to dilute the mixture, which resulted in the separation of three
distinct phases; a layer of mostly solid sulfate accumulated at the bottom of the flask, whereas the buff, water-insoluble crude product floated to
the top, leaving a pale salmon-colored suspension between the two. The mixture was vacuum filtered, and most of the salmon-buff top layer of the cake
was scraped into a beaker where it was dissolved in 10 mL of DCM. The remaining solids were transferred to a second beaker and stirred with three
consecutive 10 mL portions of DCM; the last of these was used to rinse the Büchner funnel and to extract the aqueous filtrate, which was then
extracted with one or two more such portions. The organic extracts were pooled and washed with 10 mL of 10% aqueous NaOH, which gave rise to an
emulsion that was resolved by a few additional grams of water.
The solvent was removed by first distilling the mixture under atmospheric pressure, followed by some moments under vacuum, and then by magnetically
stirring the residue under a steady discharge of argon. The oily, amber residue was cooled down close to 0°C in the hopes of observing
solidification, but there was none. 10 mL of heptane was added to give a solution with a light dusting of insolubles in it. The solid impurity was
removed via filtration through a dense ball of cotton, which was then rinsed using a dash of fresh heptane. The solution was evaporated to dryness at
room temperature, resulting in 6.67 g of a surprisingly hard, amber, crystalline solid with a phenolic, slightly woody, earthy (not moldy), medicinal,
artificial strawberry odor subtly hinting at vanilla. After melting the solid in a 50°C oven, 6.62 g remained, and had a melting point of
42.3–48.5°C
The 6.62 g of crude product was recrystallized from 19.1 g of 78.5% MeOH to obtain 5.7 g of crystals with the appearance of Himalayan rock salt and a
melting point of 51.1–52.3°C. A second recrystallization from 15.9 g of the solvent gave 5.26 g of material with less discoloration, no odor, and a
slightly higher melting point of 51.7–53.0°C; this is what was used in the subsequent formylation.
An impure residue from evaporating the mother liquors was rinsed with 3 mL of ice-cold isopropanol to afford 0.98 g of a reddish material which was
recrystallized from 2.74 g of 81% MeOH but neglected such that it evaporated nearly to dryness. Most of the impure liquor was removed by pressing the
material between folded paper towels, yielding 0.86 g of dull-brown crystals — for an unexciting total yield of no more than 45.5%, based on
phloroglucinol.
[0]: According to literature, the 1,3,5-trimethoxybenzene can be purified through steam distillation; despite reports of the neutral
C-alkylated side products also coming over with steam, it would be interesting to see the results from attempting its isolation directly from the
reaction mixture in this way.
[Fig. 12] Reaction mixture following overnight stirring

[Fig. 13] Dilution with water

[Fig. 14] Vacuum filtration

[Fig. 15] Evaporating solution of crude product in heptanes

[Fig. 16] Crystalline evaporation residue

[Fig. 17] First recrystallization

[Fig. 18] Recrystallized product

[Fig. 19] Second recrystallization
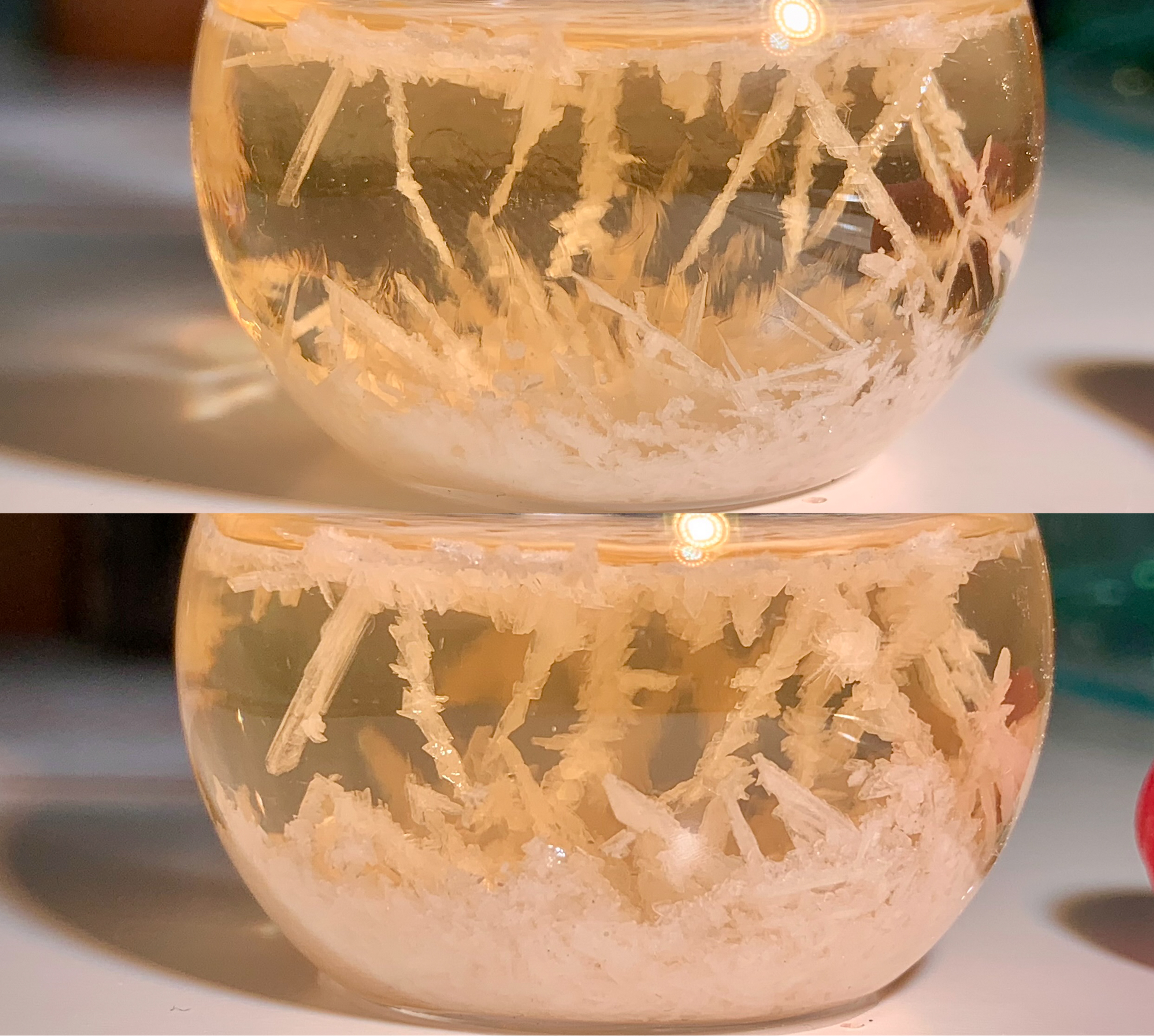
[c4] 2,4,6-trimethoxybenzaldehyde
196.20 g/mol
316.37 g/mol (bisulfite adduct; potassium salt)
A deviation from the PiHKAL procedure, this experiment substitutes N,N-dimethylformamide for N-methylformamide, because the latter is reported to
produce dramatically inferior yields of an impure product. [1] [2] Indeed, just as originally
reported by the Hyperlab member _hest, [3] this modification gave excellent results.
[1]: PiHKAL: #162 TMA-6. https://isomerdesign.com/PiHKAL/read.php?id=162
[2]: (Hyperlab) amethyst: TMA-6. https://hyperlab.info/inv/index.php?s=&act=ST&f=17&a...
[3]: (Hyperlab) _hest: Synt of 2,4,6-trimethoxybenzaldehyde. https://hyperlab.info/inv/index.php?s=1f1c32485b7603b280beee...
Experiment 1
In a round 50 mL reaction flask, 1,3,5-trimethoxybenzene (5.00 g, 29.7 mmol) was dissolved in 20 mL of DMF. A stir
bar was added, and the mixture was set up in a stationary ice bath over a stirrer. A pasteur pipette was driven through a thermometer adapter to
suspended it above the magnetically agitated mixture; through this, there was gradually injected phosphoryl trichloride (4.87g, 31.8 mmol) at a
fluctuating dropwise pace over 40 minutes. [1] Two hours after completing the addition, the
now-orange mixture was removed from the bath, whose temperature of 9°C it shared. Stirring was resumed for 2.5 hours at 24°C.
[1]: This arrangement served as an improvised way of limiting air exchange inside the flask (to protect the POCl3) in lieu
of an appropriately sized addition funnel, and worked passably: the opening was otherwise airtight, which necessitated the application of some
pressure from the dropper in hand; hence, a thin bypass through the O-ring would have been shrewd. On the other hand, the consequences of simply
alternating a stopper instead might have been negligible.
[Fig. 20] Discolored 1,3,5-TMB dissolving in DMF

[Fig. 21] Reaction apparatus

[Fig. 22] Claret hue evolving from initial additions

[Fig. 23] Reaction mixture following removal from bath

Work-up
Approximately 150 mL of cool, clean, well-stirred water from the tap was prepared in a 250 mL beaker; the contents
of the reaction flask were then added to this in <10 minutes using a dropper, causing the temperature of the mixture to rise to ~30°C before
gradually cooling down again. A sudden onset of precipitation was observed after 20 minutes. The mixture was allowed another 8 hours of stirring (in
the dark, although this is probably not important) prior to a vacuum filtration to separate the solids, which were rinsed with six 10 mL portions of
water and air-dried to obtain 5.26 grams (90.2%) of a delightfully pink, [1] crystalline solid
melting at 119.5–120.5°C.
[1]: The pink hue seems to have almost completely faded away over time, doing so much faster, initially.
[Fig. 24] Post-reaction mixture being added to water

[Fig. 25] Completed quenching mixture after some minutes of stirring

[Fig. 26] Quenching mixture after 35 minutes of stirring

[Fig. 27] Mixture prior to vacuum filtration

[Fig. 28] 1,3,5-trimethoxybenzaldehyde

This at last concludes the benzaldehyde portion of my quest for the corresponding phenethylamines!
As for what's next, I'm pleased to announce that I have made some progress on the remainder as well: Having decided (in light of some initial failures
which had fascinating implications) that it would be apt to learn more about the condensation and subsequent reduction in order to minimize the
further waste of precious starting materials, I was able to prepare all of the intended nitrostyrenes, which are currently pending reduction as I'm
figuring out the copper-catalyzed process that I quite prefer over the alternatives at my disposal. I feel like I'm finally getting close to where I
want to be, although a good deal of work still remains to be done; but I do believe that it'll all be worth it.
Whoever you are, whenever you are: thank you for making it here !
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Benignium,
Indeed, a very masterful exposition on a complicated problem. I wondered about how you were going to pull everything together. Thank you for the kind
acknowledgment. However, it is the first time that anything I have done has been called "awesome"! I am simply happy to know that I was able to help
a bit.
Your experimental write up is excellent. Good luck on your remaining synthesis pathways - keep us posted.
AvB
|
|
|
Dr.Bob
International Hazard
Posts: 2732
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
Beautiful writeup, I wish any of the papers I try to follow was as thorough, detailed and had photos. You might want to break the pieces into chunks
(maybe one post per step) for easier reading, but the photos and details are great.
|
|
|
walruslover69
Hazard to Others
Posts: 231
Registered: 21-12-2017
Member Is Offline
Mood: No Mood
|
|
Amazing Right up!
I had some some trouble with the further reduction step. Tried a few procedures that never seemed to work for unknown reasons, Eventually I found a
procedure using nickel chloride and sodium borohydride. I believe it generates nickel nanoparticles that actually act with the hydrogen to carry on
the reaction. I can dig up my procedure.
|
|
|
Quieraña
Harmless
Posts: 38
Registered: 24-8-2019
Member Is Offline
|
|
I said it before, and I'll say it again: I've read and loved pihkal and no where I have read yet that triacetoxybenzaldehyde has been used to make
triacetoxyphenethylamine, or what have you. I'm excited at the prospect of more phenethylamines that could be entheogens.
Edit: sorry, didn't know I posted about this in the same forum twice. That being said, there could be 2,5 diacetoxy, even simple acetone derivatives.
Jeez, what else can we throw in the bunch?
[Edited on 12-12-2023 by Quieraña]
Now I see there's even a thioacetoxybenzaldehyde. Crazy a whole other book waiting to be written. That substance line.. hmm.
[Edited on 12-12-2023 by Quieraña]
|
|
|
Texium
|
Thread Split
13-12-2023 at 10:34 |
| Pages:
1
2
3 |
|













