nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
OTC pyrazole synthesis
There seems to be no topic on the OTC synthesis of pyrazole yet, so with this thread I am hoping for some input regarding the best potential synthesis
route from an amateur standpoint of view, so limited glassware, equipment etc.
https://en.wikipedia.org/wiki/Pyrazole
Regardin synthesis routes; below is an exerpt from "Pyrazoles and Reduced and Condensed Pyrazoles
By Richard H. Wiley" in which multiple synthesis routes are briefly mentioned. Some seem to use exotic/toxic/dangerous intermediates (diazomethane,
N-nitrosourea derivatives), while others just seem not OTC or very amateur friendly. After some research it seems two routes seem feasible OTC:
1. Acetylacetate + hydrazine --> 3,5-dimethylpyrazole --> oxidation --> pyrazole 3,5 dicarboxylic acid --> heat (decarboxylation) -->
Pyrazole
this route seems to use the least dangerous chemicals and most OTC, though probably low yielding, especially the oxidation and likely also the
decarboxylation.
2. Patent US4434292: Acrolein (still nasty) + hydrazine (also nasty) --> pyrazoline --> chlorine (dehydrogenation) --> pyrazole
What would be the most amateur friendly synthesis? Are there better routes available that anyone knows off? Input much appreciated!

[Edited on 31-8-2017 by nitro-genes]
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
From what I remember from something I once looked up for a synthesis towards fomepizole.
Carefully controlled condensation of the sodium salt of proprionaldehyde (or in this case acetaldehyde) with methyl formate to give the salt of
alpha-methylmalondialdehyde.
Note that this requires inverse addition of aldehyde dissolved in methyl formate to a solution of sodium methoxide in methyl formate (or maybe the
other way around, anyhow a massive excess of Ester is to be present at all times. if time permits I will find the reference to this) .
Which upon treatment with Hydrazine should give the corresponding pyrazole (no reference).
Edit:
https://www.google.com/patents/WO1996016013A1?cl=en
pages 17 & 18
So OTC friendly:
Formic acid + methanol --> methyl formate
or in lieu of ice water coolers
1) Formic acid + ethanol --> Ethyl formate
2) Ethanol + nanocrystaline copper metal --(Heat)--> Acetaldehyde + H2
3) Ethanol + NaOH + Mol. Seives --> NaOEt in ethanol
All combined now:
To a solution of Ethyl formate and NaOEt in ethanol is added very slowly a solution of acetaldehyde in ethyl formate.
This should give malondialdehyde sodium salt. Neutralization of this reaction mixture with hydrazine sulfate followed by vaccuum distilation should
allow recovery of the excess ethyl formate. Later fractions will include product.
Still quite demanding in terms of equipment and the absolute requirement of anhydrous alcohol will make it hard to ever be OTC.
AFAIK Mol. Seives are not OTC.
Attachment: WO9616013A1.pdf (1.1MB)
This file has been downloaded 578 times
[Edited on 31-8-2017 by Sigmatropic]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
https://en.wikipedia.org/wiki/Ethyl_diazoacetate
1,3-cycloaddition of this with an alkene should give a pyrazole.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Why the interest in pyrazole? I have done some work on pyrazole and pyrazolone derivatives as their amino compounds form azo dyes and schiff bases
that are interesting ligands for many metals.
By the way in your Example 1 an acetoacetate ester will react with hydrazine to form 3 methylpyrazolone. If you want 3,5-dimethylpyrazole you need to
start with acetylacetone.
Another route to pyrazoles is through 4-nitropyrazole via nitromalondialdehyde which can be prepared through a complex series of reactions from
furfural via its reaction with bromine to form mucobromic acid and then nitrite salts etc. (I can post the references if you are interested).
Presumably therefore you could access pyrazole from malondialdehyde and hydrazine. Malondialdehyde is prepared in the course of the analysis of sorbic
acid and then reacting it with thiobarbituric acid form a coloured azomethine type dye. It may be possible to modify this reaction to give a
malondialdehyde solution that you can then react with hydrazine instead. The basic method is available online, check out the thiobarbituric acid
method for sorbic acid. Sorbic acid is very OTC as a wine stabilizer and fermentation inhibitor.
Sorbic acid analytical method available in:
Horwitz, W., Editor, “Official Methods of Analysis of the
Association of Official Analytical Chemists,” Thirteenth Edition,
Association of Official Analytical Chemists, Arlington,
VA, 1980, Section 20.101.
|
|
|
Dr.Bob
International Hazard
Posts: 2733
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
The synthesis of Rimonabant (the no longer sold weight loss drug) is a great example of pyrazole chemistry. The classic way to to react a diketone
with a hydrazine compound, and the pyrazole forms easily. Controlling the regiochemistry (hydazines are difunctional, so can react at either end if
substituted) is the biggest challenge, using simple hydrazine gives only one product, however. There are numerous papers on making them as drugs,
pesticides, and other useful compounds, and many ways to make them. But the simplest are 2 + 3 cyclizations and hydrazine plus a diketone type
compound. Sadly, this drugs makes you lose a lot of weight and then be depressed about it... I'm guessing it and other CB1 antonists is not
popular in this crowd... Not aware of any pyrazole agonists yet, although it would seem possible, given the number of indole agonists.
https://en.wikipedia.org/wiki/Rimonabant
[img]https://en.wikipedia.org/wiki/Rimonabant#/media/File:Rimonabant.png[/img]
http://pubs.acs.org/doi/abs/10.1021/op700110b
http://www.tandfonline.com/doi/abs/10.1080/00304948.2012.657...
http://www.sciencedirect.com/science/article/pii/S0040403908...
"Cannabinoid type-1 (CB1) receptors are promising targets for the therapeutic treatment of several neurobiological disorders. Rimonabant (Acomplia®
or Zimulti®, 1),1 a selective CB1 receptor inverse agonist, has recently been approved in the European Union (EU) for the treatment of obesity. Its
therapeutic potential may extend to the treatment of addiction2 and neurodegenerative diseases.3 At present there are only a few published routes for
its synthesis.4–7 The 1,5-diarylpyrazole core of 1 has been the subject of extensive structure–activity relationship studies to define more
strictly the key structural requirements for an improved pharmacological profile within this structural class.8
The usual method for the synthesis of 1 and its cognates employs a base-catalyzed reaction of the enolate of a substituted-propiophenone with diethyl
oxalate for 16 h (Scheme 1). The resulting diketone ester lithium enolate (2) is immediately treated with a substituted-phenylhydrazine hydrochloride
in ethanol for 20 h. The crude mixture of product hydrazones (3 and 4) are isolated by filtration, dried under vacuum and then heated at reflux in
acetic acid for 24 h, yielding a mixture of regioisomeric pyrazoles (5 and 6).5 After column purification, 6 is hydrolyzed to the corresponding
carboxylic acid (7) under basic conditions. In final steps, 1 or one of its cognates is formed by conversion of the appropriate acid into the acyl
chloride and addition of 1-aminopiperidine. Although this is an effective method for the production of several 1,5-diarylpyrazole CB1 receptor
ligands, it has several limitations. These include several steps with long reaction times, structural modification limited by the few commercially
available substituted-phenylhydrazines and lack of regioselectivity."
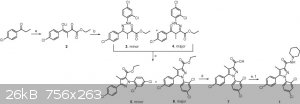
Scheme 1. Synthesis of 1. Reagents and conditions: (a) LiHMDS, Et2O, diethyl oxalate, −78 °C; (b) 2,4-dichlorophenylhydrazine HCl, EtOH; (c) AcOH,
Δ; (d) KOH, MeOH; (e) SOCl2, toluene; (f) 1-aminopiperidine, TEA, DCM.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
@sigmatropic, if this route would work it would definitely be the most amateur friendly route compared to other synthesis routes. A big if seems
whether the reaction is as efficient using acetaldehyde instead of propionaldehyde (Will try to find some more about this route). It would also
require synthesizing sodium (without molecular sieves), acetaldehyde, methylformate and even formic acid itself, which is not OTC found over here.
I've been wondering if after distillation, acrolein could immediately be reacted with (m)ethanol+sulfuric for example to yield malondialdehyde
bis(di(m)ethyl acetal) in (m)ethanol, which could be reacted further with ethanol freebased hydrazine from hydrazine sulfate. Don't like the idea of
working with a large amount of anhydrous hydrazine though, going via a diazonium salt seems more appealing. As clearly_not_atara mentioned, the
synthesis of pyrazoles seems also possible using ethyl diazoacetate (nicely OTC btw!  ), there is a section about this in the book mentioned in my first post, but haven't got much time going through these sections in detail.
Can it be as simple as paraformaldehyde + ethyl diazoacetate --> 5-carboxy pyrazole? ), there is a section about this in the book mentioned in my first post, but haven't got much time going through these sections in detail.
Can it be as simple as paraformaldehyde + ethyl diazoacetate --> 5-carboxy pyrazole? 
@boffis: The nitration of pyrazole to 4-nitro pyrazole and the reduction to 4 amino pyrazole appears to be very specific and high yielding reactions
and OTC. I was wondering if nitration of 4-amino pyrazole could produce 3,5 dinitropyrazoles or possible diazonium derivatives directly under specific
nitration conditions (was thinking maybe via rearrangement of a possible heterocyclic N-oxide?). After reading "Four Syntheses of
4-Amino-3,5-dinitropyrazole, Stefan Ek and Nikolaj V. Latypov (2014), this doesn't seem very likely though without N-protection.
[Edited on 3-9-2017 by nitro-genes]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi nitro-genes; since I have a little 3,5-dimethylpyrazole prepared some time ago I decided to try the oxidation to the dicarboxylic with alkaline
permanganate. I started out with 1.77g of 3,5-dimethylpyrazole and 11g of potassium permanganate, this is slightly less than the ideal but from past
experience of this technique a little less than the ideal amount of permanganate often significantly improves yield. I am assuming the ideal equation
is:
C<sub>5</sub>H<sub>8</sub>N<sub>2</sub> + 4KMnO<sub>4</sub> →
K<sub>2</sub>C<sub>5</sub>H<sub>2</sub>N<sub>2</sub>O<sub>4</sub> + 2KOH +
4MnO<sub>2</sub> + 2H<sub>2</sub>O
I dispersed the pyrazole derivative into 50ml of water and added 3ml of 50% KOH solution to make it alkaline and then commenced the addition of finely
ground K permanganate. Once the temperature had risen to about 60-70 C cooling was necessary and I had to add another ml of KOH solution to suppress
effervescence (presumed to be due to CO2 emmission due to over oxidation/ decarboxylation due to falling pH; note 1). After the addition was complete
the brown suspension was stirred for 20 minutes and then treated with a few ml of alcohol to ensure destruction of the permanganate. The suspension
filtered easily giving an almost clear solution. This was evaporated down slowly on a water bath to about 10ml when glistening colourless crystals
began to appear in the solution. At about 8 ml these were fairly abundant so I cooled the bowl in the fridge and filtered off the crystals. These were
not solution in dilute alkali and smelled of smoke and farmyard and are thus fairly volatile and not a carboxylic acid. The filtrate was made weakly
acid (pH 3) with 28% hydrochloric acid, causing significant effervescence, and then cooled in the fridge. Abundant colourless crystals formed
overnight, these were filtered off, washed with a very small amount of isopropanol and sucked dry. The cake was air dried at 30 C and weighed 0.93g.
(About 32% yield if it is the dicarboxylic acid)
The first crystals were found to be soluble and 2M hydrochloric acid. Just enough acid was added to dissolve then and then 2M NaOH solution was added
to precipitate the compound. The crystals formed slowly and of reasonable size and filtered easily, after drying at 30 C they weighed 0.10g (about 8%
yield).
There may be more product in the residual fluid but its getting difficult to manipulate and contains a lot of KCl now.
By the way I would be interested to see those references if you don't mind posting them here or in the "References" section.
I have run out of time again but on my return (november) I will test the melting point of eash of the compounds and the equivalence value for the acid
compound to see if it either the mono or dicarboxylic acid or a mixture. My feeling is that the reaction would benifit from lower a temperature and
that the first crystals are pyrazole itself produced by decarboxylation at the elevated temperatures giving rise to potassium carbonate as a
byproduct, hence the effervescence when acidifying the reaction mixture.
Note 1 KOH should be liberated in the reaction but in practice some is occluded by the Mn oxide and some carbonate is also formed.
Also I don't think you would need anhydrous hydrazine to condense with malondialdehyde or its acetal.
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
nitro-genes: paraformaldehyde is not an alkene, but acetylene works, giving a monosubstituted pyrazole from diazoacetate. I think vinyl bromide might
also work and it is somewhat easier to handle than acetylene. Acrylic acid could be an option as well.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
@Clearly_not_atara:
Your right, got some things mixed up probably regarding the formaldehyde.
Seems possible indeed to produce a 3-carboxy pyrazole through reaction of ethyldiazoacetate and acetylene. Maybe this artcle: "Vuluga, Daniela, et al.
"Synthesis of pyrazoles through catalyst-free cycloaddition of diazo compounds to alkynes." Green Chemistry 11.2 (2009): 156-159" could provide some
more info on the conditions utilized. This would seem fairly simple but probably depends on the solubility of the acetylene during the reaction and
efficieny of the addition reaction itself. A solvent could help, maybe ether? Acetone and MEK might react with the diazoacetate. Acrylic acid would
produce a prazole 3-one right? Not sure if this could be nitrated as well.
@Boffis
Only noticed the oxidation of 3-methylpyrazole to the carboxylic acid listed in molbase in 98% yield, it refers to some patent, which I was unable to
find so I'm not even sure if this was performed using permanganate. High pH was an issue with pyrazole IIRC, maybe due to ring opening/hydrolysis?
What is the melting point of the compound after you did the permanganate oxidation, maybe partial decarboxylation occured? Pyrazole is around 70C, any
carboxylic acid or pyrazolone would likely be higher I guess.
Posted some references in the reference section.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Acetophenones condenses with diethyl oxalate in the presence of sodium ethoxide to give benzoylpyruvate ester which should condense with hydrazine to
give a phenylpyrazole carboxylic acids. There are several patent covering such condensation. This made me wonder if there is a possibility of a
similar Claisen condensation between ethyl pyruvate and diethyl oxalate to give the sodium salt of diethyl 2,4-diketoglutarate (2,4-dioxopentandioate)
which could then be condensed with a hydrazine salts to give the 3,5-pyrazole dicarboxylate ester; hydrolysis and decaboxylation of which would give
pyrazole. This might not seem a very OTC route but sodium metal is not hard to come by and ethyl pyruvate my be accessible from Calcium pyruvate which
is available OTC as a food supplement. Oxalic acid is easily available
I was wondering if you could prepare the pyruvate ester by suspending the calcium salt in dried methanol or ethanol and pass dry HCl gas into it until
their is an excess of the latter. The calcium chloride would take up the water driving the equilibrium in favor of the ester and could then be removed
by filtration. There would be no need to isolate the ester if the alcoholic solution was dry. I have found a reference to producing ethyl pyruvate
from crude commercial pyruvic acid by salting out the solution and extracting with ether, concentration and esterification in ethanol with HCl gas.
J Amer. Chem Soc. 1944 v66(10) p1656
While trying to dig out some of my references I came across the patent attached below about the dehydrogenation of pyrazolines to pyrazoles using
sulphuric acid and iodide as a catalyst, I notice that it claims that not only does acrolein react with hydrazine hydrate to give a pyrazoline but
glycerol too! I have got to try this!! Further more it states that formaldehyde and acetone can be reacted in situ to give a hydroxyketone that acts
as a proxy form a methyl-vinyl type ketone, this presumably will give a methylpyrazole as the final product. Sounds like a messy reaction but far more
OTC than some of the above ideas
[Edited on 21-9-2017 by Boffis]
Attachment: pat4996327.pdf (272kB)
This file has been downloaded 428 times
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Brilliant if this would work, great find! A one-pot dehydration, condensation and dehydrogenation in one, and presumably no hydrazine vapours to worry
about Assuming atmospheric pressure, at 155 deg C, the mixture should be
around 66 wt% sulfuric near the end of the distilation, amazing that any condensation with the hydrazine sulfate is even possible under these
conditions. Also wonder at what temperature any acrolein is formed from dilute sulfuric and glycerol, would have guessed only at much higher
temperatures/concentration. maybe the reaction works differently and the iodine also plays a role here? Interesting
[Edited on 21-9-2017 by nitro-genes]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ nitro-genes, I must admit that as I re-read this patent and look at the laboratory preparations of acrolein from glycerol I find it hard to believe
that it would work as, like you suggest, it seems unlikely hydrazinium sulphate would react under these acid conditions unless you could simply heat
glycerol with hydrazine sulphate first (dream on ). However, I hadn't
appreciated how easy it is to prepare acrolein from glycerol either directly or via the allyl alcohol route. Maybe you could do the initial
distillation of acrolein and then add the hydrazine solution to before you acidify it and add the iodide. One point not menioned in the patent is that
much sulphur dioxide must be formed if the sulphuric acid is acting as the oxidizing agent.
Vinyl methyl ketone is prepared by the condensation of acetone and formaldehyde any way so this route sound more promising (Ullmann's -Ketones refers
to German patents 1 & 2). Does anyone know if this reaction proceeds as far as the 1-hydroxybutan-3-one stage in aqueous media. What I am getting
at is could you react 40% formalin solution with acetone to give the hydroxy ketone then react this with the hydrazine solution to get the
intermediate pyrazoline so that you don't have to isolate the rather nasty vinyl ketone. A sort of "one-pot - many stages" procedure.
German patents DT 222551 (1909) and DT 730117 (1943)
I have looked at both these patents and they are both to do with the dehydration of 1-hydroxy-3-butanone to the vinyl ketone and not with the reaction
of formaldehyde with acetone. This must be covered elsewhere.
Incidently, I notice that the formation of acrolein from glycerol is always an acid catalysed reaction. Would it be possible to use a direct basic
dehydrating agent such as calcium oxide? There is a method that uses anhydrous magnesium sulphate (JACS 1914 v36(8), pp1766-1770, ja02185a016).
[Edited on 22-9-2017 by Boffis]
[Edited on 22-9-2017 by Boffis]
|
|
|
AvBaeyer
National Hazard
Posts: 651
Registered: 25-2-2014
Location: CA
Member Is Offline
Mood: No Mood
|
|
Boffis,
You mentioned above some comments about the preparation of ethyl pyruvate. This can be prepared by the oxidation of ethyl lactate which I believe (I
am away from my library) is documented in Organic Syntheses. Ethyl lactate can be easily found from several sources as it is used by perfumery folks
among others. I think that if you try a Fischer esterification of pyruvuc acid it leads to a mixture of products.
Once in hand, ethyl pyruvate does condense with diethyl oxalate as you mentioned. Again I am unable to provide a reference at this time but I know it
exists.
AvB
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ AvBaeyer, thanks for the response, I have found a review section on the Claisen reaction on the SM library so I've got some more bedtime reading!
The reason I was looking at direct esterification of pyruvic acid is that Ca pyruvate is OTC and fairly cheap. Lactic acid is very cheap but the best
I can find is only 80% so esterification not very efficient. It may be possible to up-grade it by azeotopic distillation or a salting out/solvent
extraction method similar to that described for pyruvic acid in the JACS paper referenced above.
Any way it may be unnecessary since I have found a patent for the preparation of 1-hydroxy-3-butanone (US 2064564) which is amateur friendly and this
compound with aqueous hydrazine should offer a route to methylpyrazolinine (according to the patent I posted on the 21-sept) which I reckon can be
oxidized with alkaline permanganate directly to pyrazole-3-carboxylic acid in one step (including the dehydrogenation of the ring) and then thermal
decarboxylation to yield pyrazole. This is my route for people who don't like working with acrolein or epichlorohydrin .
Incidential nitro-genes, the method of converting pyrazolines to pyrazoles with chlorine may be unnecessary since some pyrazolines were proposed as
photographic reducing agents as they are fairly susceptible to oxidative dehydrogenation so iodine catalyzed oxidation with sulphuric acid may be over
the top and so less aggressive condition may work, say iodide and a persulphate or similar may be more practical on a lab scale. I think the chlorine
route was really being viewed as a commercial scale process.
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@nitrogenes; did you download the book; Pyrazoles and Reduced and Condensed Pyrazoles By Richard H. Wiley?
I searched on line for a download and found many site but they all seem a bit flaky.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Nope, couldn't find it either
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
This book is also known as "Chemistry of Heterocyclic Compounds: Pyrazoles, Pyrazolines, Pyrazolidines, Indazoles, and Condensed Rings, Volume 22". It
seems that with the transition to digital, the individual volumes of Chemistry of Heterocyclic Compounds have been renamed.
I have attached it for convenience (split into three files, and three posts: it won't let me attach more than 8 MB per file...)
Attachment: 1.pdf (6.8MB)
This file has been downloaded 1934 times
[Edited on 4-10-2017 by Cryolite.]
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Second post...
Attachment: 2.pdf (6.7MB)
This file has been downloaded 1218 times
|
|
|
Cryolite.
Hazard to Others
Posts: 269
Registered: 28-6-2016
Location: CA
Member Is Offline
Mood: No Mood
|
|
Final post... If a moderator is reading this please merge my files into a single post please.
Attachment: 3.pdf (6.6MB)
This file has been downloaded 777 times
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@Cryolite; very many thank for the document, most interesting and useful!
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Many thanks for the upload, also noticed some info about nitration and nitro-redutions in there, most useful!
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
While checking out some references on the preparation of pyrazole I came across a paper (1) that describes the preparation of
1-phenylpyrazole-3,5-dicarboxylic acid from the 3,5-dimethyl derivative by oxidation with potassium permanganate so it sounds like my experiment
described above was a reasonable idea. The author also describes the simultaneous 4-bromination and 3,5-de-carboxylation to yield
4-bromo-1-phenylpyrazole, I have come across simultaneous de-carboxylation and bromination before in other multi-N heterocyclic such as
1,2,3-triazole-4-carboxylic acid and 4,5-dicarboxylic acid.
An earlier paper by the same author (2) also describes the preparation of pyrazole itself from epichlorohydrin and hydrazine hydrate. This reaction
aught to give pyrazoline (a H2-pyrazole) but in actual fact pyrazole is produced directly, an excess of hydrazine hydrochloride in the reaction
mixture appears to remove the hydrogens as ammonia. Another paper by Gerhard (3) investigates this reaction with phenylhydrazine and epichlorohydrin;
aniline and ammonium chloride being the byproducts. What interests me in this reaction is the possibility that it would work with acrolein and 2 molar
equivalences of hydrazine rather than oxidizing it afterwards with chlorine. Acrolein is not pleasant stuff but if you have good quality ground joint
glassware its not difficult with well planned experiment to prepare it without gassing yourself. Or you can just buy epichlorohydrin, not exactly OTC
but obtainable.
1) Baldiano L., 1890, Berichte v23 p1448
2) Baldiano L., 1890, Berichte v23 p1103
3) Gerhard F., 1891, Berichte v24 p352
All three references are available from the French library site Galica for free. They are all in German but I may translate parts of them if people
are interested.
Edit: Actually I just read some more of the ref 1 and I see that the de-carboxylation is not spontaneous it requires heating to c 250C to get the
4-bromopyrazole deivative. When you brominate triazole dicarboxylic acids the bromine replaces the carboxylic acid group so its different.
[Edited on 11-10-2017 by Boffis]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
@ Nitrogenes, did you ever get any further with your pyrazole project?
I have found an interesting synthesis 1(2,4-dintrophenyl)pyrazole from 2,4-dinitrophenylhydrazine and malondialdehyde tetramethyl acetal in
hydrochloric acid. I also found a paper that describes the various routes to dinitropyrazoles by the unusual route of bromination then displacement of
bromine by nitration.
[Edited on 14-11-2019 by Boffis]
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Well, since nitrogenes seems to have lost interest in pyrazoles I will pursue them alone!
I have now prepared various pyrazole and pyrazolones from ethyl acetoacetate or acetyacetone and hydrazine (via its salts) and succesfully oxidized
the 3,5-dimethylpyrazole to the corresponding carboxylic acid. By neferious means I have aquired some malondialdehyde-bis(dimethylacetal) from which I
have prepared both pyrazole and 1-(2,4-dinitrophenyl)-pyrazole in high yields. I am still working on the preparartion of pyruvate and oxalate ester
though the latter is going well (see posts above for the relavance of these). I haven't yet prepared nitromalondialdehyde but I have now a reasonable
amount of bromomucic acid from which it is prepared. Interestingly my attempts to produce nitromalondialdehyde from chloromucic acid failed but did
yield some really interesting compounds.
|
|
|








