Magpie
lab constructor

Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
preparation of α-nitronaphthalene
Introduction
I used the procedure given in reference 1 to synthesize 11.8g of α-nitronapthalene. This procedure employs mixing naphthalene with acetic acid (as
solvent) then adding the nitrating acid a step at a time while controlling the temperature:
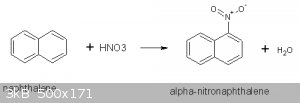
Prior to using this procedure I tried the one in Vogel (ref 2) but this turned out to be a disaster. More will be said about that under Discussion
below.
Experimental
20g of naphthalene (Enoz Old-Fashioned Mothballs) was lightly ground in a mortar. This was added to 60ml of glacial acetic acid in a 2-neck 250ml
RBF. This was then set up with an overhead mixer and thermocouple as shown below:

Synthesis of α-nitronaphthalene
A second picture shows the controls and readouts for the stepper motor mixer and the temperature indicator (°C in red):

mixer and thermocouple control/readouts
a. acid addition
The nitrating acid was made up using 30ml of con sulfuric acid (Rooto) mixed with 14ml of con nitric acid (70%). This was cooled to room temperature
then added 2ml at a time to the RBF while stirring the product at 203.5 rpm. A cooling bath was at the ready beneath the RBF but was never needed.
The temperature was to be kept at 60°-70°C. By the time all the acid had been added the temperature had just reached 70°C.
b. heating period followed by a cold water quench
The product was then kept at 70°-80°C for 30 minutes using a hot water bath. Following this the product was allowed to cool to ~40°C then dumped
into 200ml of cold water. As α-nitronaphthalene has a mp of ~58°C and a density of 1.223 it settled on the bottom of the beaker as a dark brown oil.
More will be said about this color in the Discussion below. A picture of this is shown below:

α-nitronapthalene dumped in cold water
c. grinding followed by water washing
The solidified oil was removed in large chunks and placed in a mortar for grinding:

solidified α-nitronaphthalene from oil
After grinding the product was washed on a 7cm Buchner funnel 4-5 times with cold water. It was then placed on a plate to dry overnight:

α-nitronaphthalene ground and washed
The mp of this crude product was was unacceptably low so I recrystallized it in 275ml of 90% ethanol. The recrystallized product is shown below where
you can see that the color is much improved. Ideally it would be a light yellow.

α-nitronaphthalene recrystalized
Results
After the first solidification the yield was 24.9g, or 92.1%. However the mp of 45°C, and the color, indicated that it was significantly
contaminated. Therefore a recrystallization was performed. This yielded a product with a much improved mp of 51°-53°C. However, the yield then
dropped to 11.8g, or 43.7%.
The mp for α-nitronaphthalene indicated in internet sources varies considerably from 53°-57°C (Sigma-Aldrich) to 61°-63°C (Jean-Claude Bradley).
Discussion & Conclusions
As indicated above I had earlier tried the synthesis found in Vogel's 3rd ed. This procedure specified addition of finely ground naphthalene to the
nitrating acid a little at a time followed by vigorous shaking of the RBF. This approach is faulty from at least three aspects: (1) finely grinding
smelly naphthalene is a PITA even though I used a coffee grinder instead of an open mortar, (2) the nitrating acid is always in excess, and (3) it is
difficult to monitor and control the temperature when shaking the RBF. There is significant heat of reaction.
By far the biggest problem is item (2), ie, having the nitrating acid in excess throughout the reaction. This resulted in most of my product being
dinitronaphthalene. This was confirmed by high mps of the product. The J. of Chem. Ed. procedure recognizes this liability by using a solvent and
adding the nitrating acid a little at a time. With my apparatus the temperature was easily controlled to the procedure specifications.
The only downside I can see for the J. of Chem. Ed. procedure is the formation of tar. This discolored the product and made its purification
wasteful. Still, I much prefer it over the procedure in Vogel, and it gave me the intended product.
edit:
I should note that the Vogel procedure also produced some tar. This may be an unavoidable consequence when using con sulfuric acid. Tars tend to be
taken up in oils. And there was some of this dark solidified oil produced in the Vogel procedure. I just didn't recognize at the time what this was
and discarded it.
References
1. “CONGO RED,” by E. R. Kline, Journal of Chemical Education, pp. 129-130, (1938), (see DJF90's post in this thread: http://www.sciencemadness.org/talk/viewthread.php?tid=65706#...)
2. “α-NITRONAPHTHALENE,” Textbook of Practical Organic Chemistry, by A. I. Vogel, 3rd Ed, (1958), p. 526. (see forum library)
Questions, suggestions, and comments are welcomed.
[Edited on 19-5-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Eddygp
National Hazard
Posts: 858
Registered: 31-3-2012
Location: University of York, UK
Member Is Offline
Mood: Organometallic
|
|
Very nice!
As for multiple nitration, since the nitro group is a -M group, maybe this would need a higher temperature than the mono-nitration (this definitely
happens for benzene, but probably not for naphthalene, which has a more extended conjugation that can make it more prone to have a second nitration).
Which is a shame, because monitoring temperature would have been fairly easy if it made any difference. In that case...
...as you said, adding nitric acid slowly and in stoichiometric amounts would hopefully lead to reaction with unreacted naphthalene, somewhat more
electron-rich than the product.
While 1-nitration is clearly favoured for naphthalene, is the proportion of 2-nitrated product lower than, say, 5%?
there may be bugs in gfind
[ˌɛdidʒiˈpiː] IPA pronunciation for my Username |
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Quote: Originally posted by Eddygp  |
While 1-nitration is clearly favoured for naphthalene, is the proportion of 2-nitrated product lower than, say, 5%? |
I don't think there is much dinitro product. The J. Chem Ed article says that the yield of α-nitro product is very near quantitative. My yield of
92.1% before recrystalization confirms this.
There was a minor amount of strange looking (fluffy, fibrous) light yellow by-product, however. I removed this from the supernate from the quench
water and discarded it.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
benzylchloride1
Hazard to Others
Posts: 299
Registered: 16-3-2007
Member Is Offline
Mood: Pushing the envelope of synthetic chemistry in one's basement
|
|
I ran the Vogel procedure a number of years ago and I ended up with a fair amount of dinitronaphthalene isomers which had a melting point above 200
Celsius. Any plans for the 1-nitronaphthalene? I have been doing some work with heptafluoro-2-naphthylamine, somewhat scary due to the bad reputation
of 2-napthylamine for being a carcinogen.
Amateur NMR spectroscopist
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
benzylchloride1 thanks for this confirmation of my troubles with Vogel's procedure.
Yes, I will be making napthionic acid, then Congo Red. I will avoid the scary α-naphthylamine intermediate by using the Piria reaction (ref 2 of my
OP).
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Eddygp
National Hazard
Posts: 858
Registered: 31-3-2012
Location: University of York, UK
Member Is Offline
Mood: Organometallic
|
|
I mean, before recrystallisation the "extra" yield is not 1-nitronaphthalene, because otherwise it would have neatly recrystallised with the rest of
it. Whatever it is, it has to be either initial reagents (doubtful) or some side reaction (my guesses: 1,x-dinitronaphthalene and 2-nitronaphthalene).
there may be bugs in gfind
[ˌɛdidʒiˈpiː] IPA pronunciation for my Username |
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Yes, I suppose there is some minor amount of 2-nitrophthalene. How much? I have no clue.
I attribute the loss in yield at recrystallization to α-nitronaphthalene still in the supernate.
For the extraction I heated the solution up to about 40°C (no higher to avoid oil formation) then placed the flask in ice water.
I still have the supernate and may place it in the freezer to see if I can recover more of the α-.
[Edited on 20-5-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Dr.Bob
International Hazard
Posts: 2734
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
Nice experimental. If you were running the experiment a few times, you could likely recombine all of the supernate and then concentrate to recover a
good bit more material. I have found that often getting a second crop if much tougher with only one batch. Or if you ran the remaining material
through a quick plug of silica gel, often that will all you to remove the greasy crud by washing with hexane, which might not elute the product, then
waste with ethyl acetate and you can likely elute the nitronaphthalene without all of the other more polar materials, like dinitros and polymers. I
find that often doing a quick silca gel plug gives me very nice recrystallization yields or I can sometimes crystallize most of the material, and then
recover the rest with a simple column. Just depends how valuable the material is. In many cases, 40-50% yield is fine and sufficient, if the SM
are all cheap.
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
Thanks Dr Bob for those tips on removing contaminants. I have silica gel but it is only flower shop grade. I do have some chromatography grade
aluminum oxide (neutral). Might that work as well?
I placed the supernate in my freezer (-20°C) for ~2hrs and recovered a 2nd crop as shown below. This weighed 3.9g which would increase my yield from
43.7% to 58.1%. It has an mp of 49°-50°C so is not as pure as the 1st crop. It has a somewhat darker color also I think.

The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
I did this same prep a couple of weeks ago(based on the same paper), on a 2x scale. The crude product looked essentially like dirt, so I
recrystallized a small portion from ethanol first to see how well that worked. What followed was several hours screwing around, trying various ways
of recrystallizing it to prevent it from oiling out(originally I thought that a bit of the product which was oiling out was tar), and trying to
maximize recovery. I ended up with four different products, all different colors and MPs, totaling around 35% yield, and another 10-15g of dark solid
product which oiled out and needs to be recrystallized again. I believe that methanol is a slightly better solvent than ethanol due to it's lower
boiling point, but Purification of Laboratory Chemicals 6<sup>th</sup> Edition recommends heptane or ethanol. The procedure on PrepChem is interesting, as it recommends xylene or high boiling ligroin. Seems like I've got another several hours of trying
various solvents to see.
Let me know how the Piria reaction goes. I've tried it once already with some of the crude product to get a feel for it. After two hours of
refluxing, the nitro compound had fully dissolved, so I cooled and acidified it, but nothing came out of solution. There was a substantial color
change though, to a darker red color. I've been meaning to try it again, but haven't had time as of yet, I was planning on doing it next week
sometime if I have time.
|
|
|
Eddygp
National Hazard
Posts: 858
Registered: 31-3-2012
Location: University of York, UK
Member Is Offline
Mood: Organometallic
|
|
| Quote: Originally posted by gdflp | I ended up with four different products, all different colors and MPs, totaling around 35% yield, and another 10-15g of dark solid product which oiled
out and needs to be recrystallized again.
|
Now that is unexpected!! How pure was the naphthalene that you used?
there may be bugs in gfind
[ˌɛdidʒiˈpiː] IPA pronunciation for my Username |
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
In the Kline procedure for Congo Red it says to use the solidified oil directly as precursor for the naphthionic acid. Based on this it seems that
the α-nitronaphthalene purity is not that important. But further on it states that the yield of naphthionic acid will only be in the 20-30% range.
Therefore I decided it would likely be a good plan to do some purification of the α-nitronaphthalene.
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
| Quote: Originally posted by Eddygp | | Quote: Originally posted by gdflp | I ended up with four different products, all different colors and MPs, totaling around 35% yield, and another 10-15g of dark solid product which oiled
out and needs to be recrystallized again.
|
Now that is unexpected!! How pure was the naphthalene that you used? |
I just used crushed moth balls, I didn't bother to recrystallize them(though naphthalene crystallizations are really pretty). The different colored
product was due to varying amounts of solidified oil(which is drastically darker than the crystals)/impurities in the product from the different
temperatures of recrystallization, solvents, etc.
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
A long time ago, (about 23 years ago), I did perform nitration of mothbal naphtalene with HNO3 69% only.
Thus without H2SO4.
I did put the ball into a test tube, and I added about 4 times the volume of HNO3 69%.
The mothbal was floating on top (test tube was thus about half full)
Then I allowed a common 75W lightbulb (yes one of those old tungsten wire ones) to heat it up from above by holding the tube close to the lamp (with a
woden clamp).
When molten the naphtalene was a clear transparent liquid (so T° was above 80°C (the mp of Naphtalene) but below the boiling point of 69% HNO3).
I did put a cap on top when hot to avoid too much smell of the sublimating naphtalene.
While heating the HNO3 was turning the clear phase above it yellow...then after a good while (1 hour), it turned deeper yellow.
I let the all test tube cool down and repeated the heating of the same test tube from time to time one day on two days, then cooled again.
After about 3 treatments (one week --> about 3 hours), the solid was nearly orange and had a very distinctive smell totaly different from initial
naphtalene...from which smell had disapeared (or was covered/masked).
This orange waxy solid was heavier than the initial naphtalene...almost sinking into the used HNO3.
It was very different solid...with a different kind of crystalinity so to speak.
It was washed with boiling water and hot bicarbonate water and dried then used with KClO3/NaClO3 for Cheddite experiments.
What is interesting here is that just like for my toluen experiment (yielding nitrotoluen and nitrobenzoic acid), no H2SO4 nor concentrated HNO3 was
used ...only 69% azeotropic HNO3!
Nothing else could have been formed since only mononitration can occur in such media and naphtalene is like benzene quite resistant to oxydation (less
than C6H6 of course but more than C6H5-CH3).
Sadly I didn't perform at that time a sublimation test to be sure there was no more naphtalene present inthere nor did I made a melting point testing.
If I have time and no ex-girlfriend at home (sadly can take monthes), I want to redo that fast experiment and confirm those old datas with more
detailled results.
[Edited on 8-6-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
The napthalene mothballs have finally arrived !
Let's see if i can replicate Magpie's experiment.
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Sorry for the long delay - I've been meaning to comment on this for while now but haven't had the opportunity to find sufficient time to write a
comprehensive and coherent reply.
| Quote: Originally posted by Magpie |
The nitrating acid was made up using 30ml of con sulfuric acid (Rooto) mixed with 14ml of con nitric acid (70%). This was cooled to room temperature
then added 2ml at a time to the RBF while stirring the product at 203.5 rpm. A cooling bath was at the ready beneath the RBF but was never needed. The
temperature was to be kept at 60°-70°C. By the time all the acid had been added the temperature had just reached 70°C.
|
I have two trains of thought on this, but both involve dropwise addition of the nitrating acid rather than portionwise addition. The first idea is to
perform the addition with cooling in an ice bath. This may minimise/eliminate the formation of tarry material, but may allow for accumulation of
nitrating acid and potential (although unlikely) exothermic runaway when cooling is removed. This leads me to the second idea - to pre-heat the
naphthalene suspension/solution before dropwise addition of nitrating acid (further comment on this later). I'm not sure if/how this might affect the
formation of tarry material.
| Quote: Originally posted by Magpie |
The product was then kept at 70°-80°C for 30 minutes using a hot water bath. Following this the product was allowed to cool to ~40°C then dumped
into 200ml of cold water. As α-nitronaphthalene has a mp of ~58°C and a density of 1.223 it settled on the bottom of the beaker as a dark brown oil.
|
Did you check for the complete consumption of starting material before quenching the reaction? I'm not sure if you have access to TLC plates, but I
think that would be the easiest and most appropriate way. You say that you had a dark brown oil separate out after drowning the reaction mixture in
water, but make no mention of solidification on cooling? I assume this was an accidental omission, but should be added for clarity.
| Quote: Originally posted by Magpie |
After grinding the product was washed on a 7cm Buchner funnel 4-5 times with cold water. It was then placed on a plate to dry overnight
|
Given that you have access to mechanical stirring, you might consider adopting the workup provided on PrepChem (melting the crude product under water,
agitating, allowing to settle and cool, then decant the aqueous supernatant and repeat). I suspect you'll find it more efficient at removing residual
acids compared to washing on the filter, but I appreciate it is more involved. If you confirm complete consumption of naphthalene prior to quenching,
then you can omit the steam distillation. I would certainly advice recrystallisation; more of that below.
| Quote: Originally posted by Magpie |
The recrystallized product is shown below where you can see that the color is much improved. Ideally it would be a light yellow.
...yielded a product with a much improved mp of 51°-53°C. However, the yield then dropped to 11.8g, or 43.7%. The mp for α-nitronaphthalene
indicated in internet sources varies considerably from 53°-57°C (Sigma-Aldrich) to 61°-63°C (Jean-Claude Bradley).
|
I'd recommend including a carbon treatment into the recrystallisation from ethanol. You should observe a suitable improvement in the colour of your
product. As the product is a low-melting solid, I'd dissolve it in minimal solvent around the expected melting point (should minimise/prevent oiling
out), add 0.1 wt of activated carbon, heat to reflux for a short period and filter whilst still hot. As the solution cools to saturation point (around
the expected meltng point) you should observe crystallisation rather than oiling out. You may also consider the use of aqueous alcohol to improve the
recovery of the material, assuming solubility in water is lower than that in ethanol. You may also consider the use of isopropanol as a substitute for
ethanol, either with or without the presence of water.
The difference in literature values might be explained by different polymorphs. You might be able to check this by looking at the crystal structures
under a microscope (e.g. platelets, needles, prisms). Sometimes it is found that the use of a different solvent for recrystallisation affords a
different polymorph. It may be that the lower value you have observed is obtained when ethanol is used as solvent, but the higher value (as mentioned
on the PrepChem page) is observed when xylenes or ligroin is used. It certainly wouldn't surprise me if this was the case. When you're making a
melting point determination, you're really looking for the sharpness of the transition as opposed to the actual value, which can depend on rate of
heating, accuracy of thermometer etc. That being said, there are some compounds known to give broad, poorly-defined melting points, but I don't think
this is one of them.
| Quote: Originally posted by Magpie |
...finely grinding smelly naphthalene is a PITA even though I used a coffee grinder instead of an open mortar...
|
I'd address this issue by recrystallising the mothballs. This will ensure that the naphthalene is of good purity before use, and also provides a
finely divided material (assuming you stir during cooling). Alternatively, if the reaction is preheated before addition of the nitrating acid then the
solubility of naphthalene should be high enough to afford full dissolution (wikipedia states 111g/100g AcOH at 60 *C)
A final point to address is one that PHILOU Zrealone has brought up - it might not even be necessary to use sulfuric acid and it may afford a cleaner
reaction/less tarry material. Certainly worth a go on small scale, and I'd probably recommend employing the "preheated method" that I suggested above
in this case.
[Edited on 28-6-2016 by DJF90]
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
| Quote: Originally posted by DJF90 |
Did you check for the complete consumption of starting material before quenching the reaction? I'm not sure if you have access to TLC plates, but I
think that would be the easiest and most appropriate way. You say that you had a dark brown oil separate out after drowning the reaction mixture in
water, but make no mention of solidification on cooling? I assume this was an accidental omission, but should be added for clarity.
|
I don't have TLC plates.
The oil did solidify, as recorded in my notebook.
| Quote: Originally posted by Magpie |
The recrystallized product is shown below where you can see that the color is much improved. Ideally it would be a light yellow.
|
| Quote: Originally posted by DJF90 |
I'd recommend including a carbon treatment into the recrystallisation from ethanol. You should observe a suitable improvement in the colour of your
product. As the product is a low-melting solid, I'd dissolve it in minimal solvent around the expected melting point (should minimise/prevent oiling
out), add 0.1 wt of activated carbon, heat to reflux for a short period and filter whilst still hot. As the solution cools to saturation point (around
the expected meltng point) you should observe crystallisation rather than oiling out. You may also consider the use of aqueous alcohol to improve the
recovery of the material, assuming solubility in water is lower than that in ethanol. You may also consider the use of isopropanol as a substitute for
ethanol, either with or without the presence of water.
|
I agree that efforts to obtain a pure product here are worthwhile as long as yield is acceptable.
| Quote: Originally posted by Magpie |
...finely grinding smelly naphthalene is a PITA even though I used a coffee grinder instead of an open mortar...
|
| Quote: Originally posted by DJF90 |
I'd address this issue by recrystallising the mothballs. This will ensure that the naphthalene is of good purity before use, and also provides a
finely divided material (assuming you stir during cooling). Alternatively, if the reaction is preheated before addition of the nitrating acid then the
solubility of naphthalene should be high enough to afford full dissolution (wikipedia states 111g/100g AcOH at 60 *C) |
Enoz claims 99.95% purity and the mothballs are dazzling white crystals.
[Edited on 28-6-2016 by Magpie]
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
Fery
International Hazard
Posts: 1016
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Online
|
|
Recently I performed this nitration and tried to minimize acetic acid use (I used only 1/3 of the amount) and also maximize nitric acid use (I did not
use HNO3 excess like 0,20 mol HNO3 for 0,15 mol of naphthalene as the excess could lead into dinitration although low temperature may effectively
prevent it)
H2SO4 is present to utilize all the precious HNO3. If using only HNO3 without H2SO4 substantial amount of HNO3 will stay unreacted.
I will report more after the workup is done (only the nitration part is already done).
Meanwhile I would like to share my theoretical thoughts and some calculations.
I performed the synthesis at 1 mol scale.
1,00 mol naphthalene (128.2 g) + 1,00 mol HNO3 (97,0 g 65% = 70 ml) + 150 ml 96% H2SO4.
I used only 150 ml of glacial acetic acid as a solvent (instead of suggested 60 ml for 20 g of naphthalene) in which even after dilution with water
(from nitration mixture and reaction water) enough naphthalene is certainly dissolved at every moment to be reacted with nitration mixture from
dropping funnel. There is only 18 g of reaction water = 1 mol and 34 g = 2 mols of H2O from 65% HNO3 which is bound by 275 g H2SO4 = 2,8 mol.
Separation of possible unreacted naphthalene seems to be trivial by steam distillation. Separation of dinitro from 1-nitro by crystallization may be
more challenging (oiling out).
Here calculations of steam distillation.
Info obtained from here:
https://sci-hub.ee/10.1021/acs.est.6b06364
For sake of simplicity assume only binaries during steam distillation: water + naphthalene, water + 1-nitronaphthalene (in reality it will be ternary
H2O + naphthalene + 1-nitronaphthalene)
log10 P = A - (B/(C+T))
P is the saturated vapor pressure in kilopascals (kPa) and T is the temperature in kelvins.
1-Nitronaphthalene
Antoine III (liq) A = 7.8959, B = 3468.4, C = 0, dev = above 5.0, range = 332 to 580
Naphthalene
Antoine IV (liq) A = 6.19487, B = 1782.509, C = -65.637, dev = 0.1, range = 352 to 500
using the Antoine equation and constants the vapor pressure of naphthalene at 373 K is P = 2.486 kPa so the water vapor pressure is 98.8 kPa (so the
water will distill at a temperature little lower that 373 K but that could be neglected)
so the distillate contains 2.5*128.2 = 320.5 g naphthalene with 98.8*18 = 1778.4 g of water
1-nitronaphthalene vapor partial pressure 0.03956 kPa at 373 K and H2O partial pressure 101.3 kPa
so the distillate contains 0.03956*173.2 = 6.852 g of 1-nitronaphthalene with 101.3*18 = 1823.4 g of water
if I assume that the nitration did not proceed quantitatively and there is 32 g of unreacted naphthalene (25% of the reactant due to side reactions
wasting the HNO3 or according the fact that I did not use HNO3 in excess) that would require 178 g of water for distilling it out and in such amount
of steam 6.85 * 178 / 1823 = 0,67 g of 1-nitronaphthalene is distilled out
during steam distillation loses of 1-nitronaphthalene are acceptable low and such a naphthalene with a little of 1-nitronaphthalene
could be used in repeated 1-nitronaphthalene synthesis, by my opinion having unreacted naphthalene in the crude nitration product is much easier
situation to deal with than having there dinitronaphthalene
so a steam distillation will be certainly a part of my workup procedure to remove unreacted naphthalene as I used reactants in 1,00:1,00 molar ratio
instead of the ratio 0,15 mol naphthalene (20 g) : 0,20 mol HNO3 (14 ml 65% HNO3)
even distilling 100 ml more water after all naphthalene is distilled out would lose only less than 0,5 g of the product (that would ensure that
unreacted naphthalene is separated out even more thoroughly)
also the point when naphthalene stops to distill out with steam should be well visible as the amount of aromatic compound in the condenser should drop
approximately 50 times
I'm also missing a washing step with something like 1-2 % NaOH or at least Na2CO3 or at least NaHCO3 to remove acids, but maybe repeated washing with
hot water is sufficient. I will perform such a washing step in my workup certainly. There could be also some phenolic compounds (1-naphthol,
2-naphthol, naphthoquinones etc, look at the dark color of the crude nitrated product) as a side reaction products of oxidation so alkaline washing
(NaOH or Na2CO3) should remove them in a form of water soluble naphtholates.
[Edited on 11-9-2021 by Fery]
|
|
|
vibbzlab
Hazard to Others
Posts: 241
Registered: 6-11-2019
Member Is Offline
Mood: Always curious
|
|
I would love to hear how the prep was done. I mean did you add the acid in one portion or did you add the acid dropwise , how long did you go with
nitration and regarding the ice bath
Amateur chemist. Doctor by profession
Have a small cute home chemistry lab.

Please do check out my lab in YouTube link below
This is my YouTube channel |
|
|
vibbzlab
Hazard to Others
Posts: 241
Registered: 6-11-2019
Member Is Offline
Mood: Always curious
|
|
Thanks to magpie and his notes , I was successfull in attempting the synthesis of 1-nitronaphthalene and got exactly the same yield as he did. This
video is also a tribute to his efforts
https://youtu.be/oRN9ExHEdcY

Amateur chemist. Doctor by profession
Have a small cute home chemistry lab.
Please do check out my lab in YouTube link below
This is my YouTube channel |
|
|










