byko3y
National Hazard

Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
HI+P reduction mechanism
I've already spent a lot of time trying to understand the mechanism of reduction of benzyl alcohols and other compounds.
Some say it's all about reversible reaction
H2+I2 <-> 2HI
but it doesn't seem to play important role in the reduction, mainly because you can't reduce CH3I to methanol using HI.
There's few articles about the reduction, such as:
- "Reduction of benzylic alcohols and α-hydroxycarbonyl compounds by hydriodic acid in a biphasic reaction medium" http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3302097/ (free acess)
A lot of examples of P+HI system, plus description of iodine regeneration cycles.
- "Further studies on the reduction of benzylic alcohols by hypophosphorous acid/iodine" http://quod.lib.umich.edu/a/ark/5550190.0006.634/1 (free access)
Also describes some interesting examples of vinyl phenone reduction, leading to polimeric products mainly and 20% of terminal iodide
(3-phenylpropyl-1-iodide).
- "Reduction of diaryl alkenes by hypophosphorous acid–iodine in acetic acid" http://www.sciencedirect.com/science/article/pii/S0040402002...
Authors perform aryl olefins reduction, also they try to use acetic anhdyride as a medium, this way obtaining acyl derivatives as a byproduct, try
to explain the pattern of reactions.
- There's also "Hypophosphorous acid–iodine - novel reducing system" articles, part 1 and part 2, but I found no usefull information there.
Some examples of reactions I've found in "e-EROS Encyclopedia of Reagents for Organic Synthesis", article about hydrogen iodide:
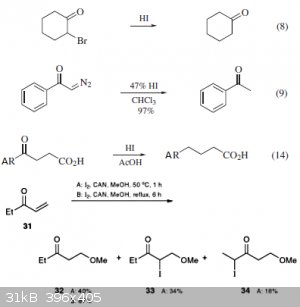
The last one is taken from google books, "Science of Synthesis: Houben-Weyl Methods of Molecular Transformations Vol. 37: Ethers", p. 103 -
Iodomethoxylation of unsaturated ketone via I2 + NH4 Ce NO3 + MeOH, then reduction after a long time.
Hudlicky in his "Reductions in Organic Chemistry", p.53 mentiones that for really reactive α-bromoketones and α-bromothiophene it's possible to
reduce them even with hydrogen bromide.
What do those compounds (ketones, benzyl alcohols, benzyl halides, styrenes) have in common? I think it's all about capability of addition of
electrophile. Ketones undergo aldol condensation and mannich reaction by adding a positively charged iminium or aldehyde, but the ketones are much
more reactive at alpha position. Benzyls can be treat as a less reactive analog of ketone. They can be halogenated and benzylic position just like
ketones, but for aldol condensation you need an ortho-electron withdrawing group: o-nitrotoluene undergoes a condensation with formaldehyde yielding
2-(2-nitro-phenyl)-ethanol ).
So I'd say it's all about nucleophility of the compound. But is there another example of a similar nucleophilic coumounds, that can be reduced by
action of hydrogen halide? I'm pretty sure I've found them. They are chloro- and bromoamines. Just like carbon and iodine have relative
electronegativity of 2.5, nitrogen and chlorine have electronegativity of 3.0 (bromine 2.8, but it is pretty close too).
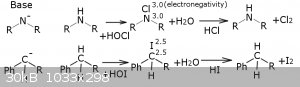
At the picture you can see the equivalent reactions that can be performed with those compounds. Probably I could use experimental data of chloramines
to understand behavior of benzyl halides.
The main question that bothers me for many time is: what will happen to this compound after reduction with P+I?

|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I see no answers, so I'm gonna give some my ideas based on reasearch on another similar compound:
  
Regular halogen reduction products. I'm not sure about favorable double bound position.

Sic! Alkylation is also comfirmed by the article I've already mentioned http://quod.lib.umich.edu/a/ark/5550190.0006.634/1

Full reduction product.
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Since you're still pretty active and never got an actual response, I'll give it a go. Like you, I spent a lot of time trying to understand this
reaction. The reduction of benzyl alcohols with HI may be an old and established reaction, but when it comes to specific details regarding the actual
reduction mechanism itself, there seems to be very little conclusive literature to be found. You'd think with how popular it became after the meth
cooks discovered it that there would be a bit more out there on its mechanism. Between the HI/P and equally-elusive Birch-style reductions of benzyl
alcohols, I've nearly driven myself crazy trying to figure out how they work.
The good news is that, after years of hair-pulling, I'm pretty sure I've finally figured out how these damn reductions work. Here's my take on the
HI/P mechanism:
Quote: Originally posted by byko3y  | Some say it's all about reversible reaction
H2+I2 <-> 2HI
but it doesn't seem to play important role in the reduction, mainly because you can't reduce CH3I to methanol using HI. |
You're right about it having nothing to do with the H2 + I2 ⇌ 2 HI equilibrium that many have assumed. I believe the real key
to this reaction is actually a radical carbon intermediate. The reduction involves homolytic cleavage of the C-I bond to give a radical carbon,
followed by a single-electron transfer from a nearby iodide anion to give a carbanion. The two iodine radicals then combine to form I2, and
the carbanion grabs a proton off of hydronium.
Keep in mind that, due to iodine's size, not only does it form extremely weak bonds with carbon, but the iodide anion also has a tendency to lose a
valence electron and become a radical. I believe this to be the key to HI's reducing power. A radical intermediate is also suggested in your first
reference, by the way. (I've read it many times in the past) Also, if you check out page 537 of that book you referenced (Science of Synthesis:
Houben-Weyl Methods of Molecular Transformations), there's actually a brief section on homolytic cleavage of C-I bonds to give radicals.
| Quote: | | What do those compounds (ketones, benzyl alcohols, benzyl halides, styrenes) have in common? |
Think about it. What do every single one of those compounds have in common that non-reducible iodomethane does not? They all have a π-system next to
the carbon being reduced. In other words, they all have a functional group that could potentially stabilize an adjacent radical carbon. The benzylic
carbon of a benzyl alcohol, benzyl halide or styrene can be stabilized by the nearby phenyl ring, and the hydroxyl-group carbon of an
α-hydroxycarbonyl compound can be stabilized by the nearby carbonyl group. It is the stability of that radical intermediate that allows the
molecule to be reduced by HI.
Here's the mechanism I'm proposing. I drew this in ChemDraw a while back; however, there's a second possible mechanism that I did not bother to draw,
which is one where no C-I bond is ever formed. Instead, the iodide anion in the second step just transfers an electron to the benzylic carbocation to
give the benzylic radical in step four. The rest of the reaction would proceed as shown, though.

As far as Birch-style reductions of benzyl alcohols go, I believe they also hinge on a benzylic radical intermediate. Basically, the first electron is
transferred into the phenyl ring to give a radical anion. A pair of electrons then form an exocyclic double bond between the ipso-carbon and
the benzylic carbon, cleaving the hydroxyl group as lithium hydroxide. The resonance-stabilized benzylic radical then forms, which undergoes a second
electron transfer to give a carbanion, followed by abstraction of a proton from a proton source.

[Edited on 9-10-2015 by Darkstar]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
byko,your question reminds me of the synthesis of krokodil from codeine by the HI/red P method.
the codeine is treated with HI/red P to break the phenolic ether and simultaneously reduce the allyl alcohol in another ring in the codeine
molecule(reduction of the 6-hydroxyl group along with the 7,8 double bonds.
I always had a doubt why the other ether linkage wouldn't break as well
https://en.wikipedia.org/wiki/Desomorphine
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Same kind of weird effect is seen with glycerol (propan-1,2,3-triol) and HI...
One would expect 1,2,3-triiodopropane; because when HBr or HCl are used, tribromo or trichloro compound are obtained; but with HI you end up with
allyl iodide (CH2=CH-CH2I + I2)...
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by CuReUS | byko,your question reminds me of the synthesis of krokodil from codeine by the HI/red P method.
the codeine is treated with HI/red P to break the phenolic ether and simultaneously reduce the allyl alcohol in another ring in the codeine
molecule(reduction of the 6-hydroxyl group along with the 7,8 double bonds.
I always had a doubt why the other ether linkage wouldn't break as well
https://en.wikipedia.org/wiki/Desomorphine |
It probably does break the second ether linkage to some extent; however, it's probably not all that favorable without a ring rearrangement
(more details below) since it would create a secondary carbocation. And while I'm far from an expert on the reaction, from what I understand, the
reduction of codeine to desomorphine via the HI/P method isn't exactly clean like it is with ephedrine/pseudoephedrine to methamphetamine. Unlike the
latter, the reduction of codeine usually ends up producing all kinds of additional, unwanted crap. In addition to all the various iodonated,
methylated and partially-reduced byproducts that using HI likely generates, I'd imagine it also produces apomorphine as well via dehydration and rearrangement of the C ring, followed by protonation and subsequent cleavage of the second ether linkage to give a more stable tertiary carbocation.
With that said, HI does seem to reduce codeine to desomorphine as well, so clearly it's able to reduce both the 6-position hydroxyl group as well as
the 7,8-double bond. If we assume that this is true, then the reduction is actually quite interesting from a mechanistic point of view, particularly
the reduction of the double bond. Reduction of the hydroxyl group is easy if you believe the mechanism I proposed in my other post. The π-system of
the double bond would easily stabilize the adjacent radical carbon, so that's how the hydroxyl group is able to be reduced. The reduction of the
double bond, on the other hand, isn't so clear. My guess is that it's first protonated, adding a hydrogen to the 7-position carbon and creating a
secondary carbocation at the 8-position. Since the adjacent carbon (position-14) is tertiary, the molecule immediate undergoes a 1,2-hydride shift to
give the more stable tertiary carbocation. The carbocation is then iodonated followed by homolytic cleavage of the C-I bond to give a relatively
stable tertiary radical. The radical is then reduced to a carbanion via single-electron transfer from iodide, which then abstracts a proton from
hydronium.
And while it's probably not as likely as the above, there's also a second possible mechanism that I thought of where the hydroxyl group is protonated
and then eliminated via rearrangement of the C ring similar to how apomorphine forms (see second and third step in the Wiki link), which creates two double bonds right next to one another. The first
double bond is able to be reduced because the second double bond stabilizes the radical carbon that results, and the second double bond is reduced
because it's able to form a stable tertiary radical carbon at position-14. The double bonds are ultimately reduced just like before via protonation
--> iodonation of carbocation --> homolytic cleavage of C-I bond --> single-electron transfer from iodide.
Anyway, I had some free time (I really just love playing around in ChemDraw  )
so I drew the first mechanism I proposed. Of the two, I think it's the more plausible. I figured that the reduction mechanism for codeine might also
more directly answer byko3y's question(s), too, which is another reason I drew it. Of course, I could be missing something and end up being completely
wrong, but then again that's what makes trying to figure out mechanisms so much fun. So what do you think, CuReUS, since you brought it up? What about
the rest of you? I'm always open to any input I can get, especially from people as knowledgeable of those on SM. )
so I drew the first mechanism I proposed. Of the two, I think it's the more plausible. I figured that the reduction mechanism for codeine might also
more directly answer byko3y's question(s), too, which is another reason I drew it. Of course, I could be missing something and end up being completely
wrong, but then again that's what makes trying to figure out mechanisms so much fun. So what do you think, CuReUS, since you brought it up? What about
the rest of you? I'm always open to any input I can get, especially from people as knowledgeable of those on SM.
EDIT: added the missing HI between steps four and five.

[Edited on 9-30-2015 by Darkstar]
|
|
|
byko3y
National Hazard
Posts: 721
Registered: 16-3-2015
Member Is Offline
Mood: dooM
|
|
I have no inspiration to interpret the information, so I post it as is.
HBr actualy is capable of reducing carbanions
The Hydride Transfer Nature of the Reduction of Carbonium Ions by HBr, HI and a Pt and an Ir Hydride
HI can reduce alkyl iodides, so the iodination reaction is truly reversible.
Kinetics of the Thermal Reaction of Gaseous Alkyl Iodides with Hydrogen Iodide
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
How do you expect to ever figure out these mechanisms if you don't bother to read the relevant literature? There's no clear-cut answer or general
consensus on how these reductions work, so all anyone can really do is read the available literature and decide for themselves.
Carbanions (R3C−) have a lone pair of electrons and a negative charge. Since they have a full octet and are bases, they can just grab a
proton off of something. There's no need to reduce them. Did you mean carbocations, more specifically of the carbenium (R3C+)
variety? Because that's what the paper is referring to. (although the paper, being over 50 years old, is actually using the outdated "carbonium"
nomenclature, which technically refers to something completely different nowadays)
Positively-charged carbenium ions are trivalent and have an empty orbital. The orbital could either be filled/reduced by a pair of electrons via
nucleophilic attack:
R3C+ + Nu− ---> R3C−Nu
or by one electron at a time through some sort of SET mechanism:
R3C+ + e− ---> R3C* (radical)
R3C* + e− ---> R3C−
The resulting carbanion can then grab a proton or some other electrophile.
Like I've said, I think HI is unique and completely unlike the other hydrohalic acids because of iodine's large size and just how easily iodide anions
can be oxidized. I also believe iodine's size to be the key to HI's reducing power, too. It also explains the unique behavior and relative instability
of alkyl iodides compared to the other alkyl halides, as C-I bonds are typically rather weak and easily cleaved homolytically to give radicals. Why do
you think alkyl iodides like iodomethane need to be stored in the dark to prevent decomposition and iodine formation? Also, the relative ease of
reduction when an adjacent π-system is present also strongly suggests some sort of radical mechanism for reasons mentioned in my first post. In fact,
radical intermediates are actually suggested in the references in your original post, and in the first one they even use TEMPO to try and trap them.
Bottom line: I believe HI reductions more than likely involve a radical carbon followed by an SET from iodide. Whether the HBr reductions work this
way as well, I don't know. I just hope that this thread has been of at least some value to you.
[Edited on 9-30-2015 by Darkstar]
|
|
|
|








