Joeychemist
Hazard to Others

Posts: 275
Registered: 16-9-2004
Location: Canada
Member Is Offline
Mood: Sedated
|
|
PYX
What dose any one know about PYX? Dose any one have any experience with this explosive?
I have searched the MSDB files and found no mention of this explosive or its procedure. This was written by Megalomania from various references and
sources, but I have re-typed it, to bring to this forum for discussion. This was copied from his ruff copy, I’m not sure if he has finished it yet
or not. So here is what we have to start with;
PYX is a insensitive high explosive (2,6-Bis(picrylamino)-3,5-dinitropyridine)
Mp 360 C
Density 1.77 g/mL
VoD 7448 m/s
C17H7N11O16
From what I’ve read the procedure would go like this;
Prepare a solution of 100ml water and 10.9 g of 2,6-diaminopyridine In a glass beaker, Then In a 1000-mL round-bottom Florence flask, prepare a
solution of 400 g of isopropyl alcohol (99.9%) and 55 g of picryl chloride. Then stir the solution well to dissolve some of the chloride and add 8.4 g
of magnesium carbonate. Heat both the beaker and the flask to 28°C while stirring. Continue stirring until the picryl chloride completely dissolves
in the alcohol and the diaminopyridine is mixed with the water.
The next step is to configure the flask for addition and reflux. Add the water-diaminopyridine mix to the flask of alcohol solution slowly over a 30
minute period while maintaining a gentle reflux. The addition will generate some CO2 waste, so control the addition accordingly to regulate the
reaction. After the addition, continue refluxing for about 5 hours. At the end of the refluxing period, pour the hot solution over a filter to collect
the crystals of the intermediate compound 2,6-bis(picrylamino)pyridine. Wash this compound several times with small portions of water. Yield should be
about 95-98%.
The next step is to Add 825 parts by weight of 98% nitric acid to a 1000-mL round-bottom Florence flask. Slowly add 100 parts by weight of wet
2,6-bis(picrylamino)pyridine as prepared above to the flask at such a rate as to keep the acid between 0°C and 30°C. Having an ice water bath handy
may help if the temperature rises too much. After the addition, set the flask up for reflux and heat the flask to the reflux temperature of the nitric
acid (about 76°C) while stirring. Continue to reflux the flask for about 5 hours. Afterwards, discontinue cooling the reflux condenser and heat the
flask (about 84°C) to distill off about 20% of the nitric acid.
Allow the acid mix to cool to about 27°C and the solid product to settle. Decant off as much acid as possible and vacuum filter the remaining
contents to collect the solid product. An acid resistant filter paper is advised, or the acid can be diluted with a large quantity of water before
pouring on the filter paper. Wash the obtained product of PYX several times with distilled water to remove all traces of acid. The wet product is
placed in an oven and heated at 38 C until a constant weight is obtained.
Optionally, PYX of increased sensitivity can be prepared. As PYX is rather difficult to detonate reliably, it is sometimes advantageous to make it
more sensitive. Into a small beaker add 1 part by weight (for example 50 g) of PYX prepared above to 5 parts by weight (about 250mL) of DMSO (can be
purchased OTC). Heat the beaker to 80-85°C while stirring until all of the PYX dissolves. Slowly pour the hot solution into another beaker containing
100 parts by weight (4 L) of acetone. It is important to add the solution to the acetone, not the acetone to the solution. Stir the mixture slightly
to mix it.
The hardest things to find for your regular Joe would be the 2,6-diaminopyridine and picryl chloride. These will be very tricky for some.
Thats what we have to go on, Lets see you guys run with it.
Merry Christmas
[Edited on 24-12-2004 by Joeychemist]
|
|
|
IPN
Hazard to Others
Posts: 156
Registered: 31-5-2003
Location: Finland
Member Is Offline
Mood: oxidized
|
|
I was planning on making some of it. I even got my hands on some of the 2,6-diaminopyridine, but I couldn't find any easy way to make the
required picryl chloride.
Though I have few hundred grams of white phosphorus I don't know if I want to try to make POCl3 from it. Phosphorus is a nasty chemical. 
|
|
|
Nick F
Hazard to Others
Posts: 439
Registered: 7-9-2002
Member Is Offline
Mood: No Mood
|
|
Picryl chloride:
2,4,6-trinitroaniline --(diazotise)--> 2,4,6-trinitrobenzenediazonium anion --(Sandmeyer rxn)--> picryl chloride
TNA can be prepared by nitrating aniline carefully without a large excess of sulphuric acid, though this is not likely to be an easy reaction to
control. Alternatively, you could deactivate aniline first by forming the acetamide with acetic anhydride, nitrate that, and then hydrolyse the
product. This is likely to be a better yielding reaction. Nitration of aniline can yield 2,3,4,6-tetranitroaniline (even in one step), treatment of
this with NaN3 in ethanol will give an aminodinitrobenzofuroxan which, assuming nitrous acid doesn't mess it up (which it could...), might
produce a nice cousin of PYX in the later stages. I'm too tired to think about what HNO2 might do to a benzofuroxan, but I wouldn't rule out
unwanted reactions without doing so.
I believe aniline can be prepared by heating benzoic acid (easy to get) with urea (easy to get), followed by a Hoffman degradation with sodium
hypochlorite (easy to get). Or, possibly, though I still haven't got round to trying it, TNA may be prepared by nitrating benzoic acid to m-nitro
(~80% yield, along with some o- and p-nitro), reducing this to m-aminobenzoic acid (Zn/HCl, should be an OK yield - nitrobenzene is reduced to aniline
almost quantitively), trinitrating that (should be a nice enough reaction) to aminotrinitrobenzoic acid, and decarboxylating that (which should happen
easily in boiling water). Et voila, OTC TNA, possibly . I really want to try this
soon, I have no excuse for not doing so, except that I'm really lazy.
I have no idea how to make 2,6-diaminopyridine though...
|
|
|
Turel
Hazard to Others
Posts: 141
Registered: 29-11-2003
Location: The Hardware/Software Interface
Member Is Offline
Mood: Thixotropic
|
|
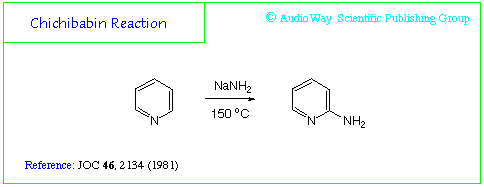
Chichibabin Reaction
Could be a good start.
|
|
|
Nick F
Hazard to Others
Posts: 439
Registered: 7-9-2002
Member Is Offline
Mood: No Mood
|
|
Yeah, that should work. Now, OTC pyridine and sodamide?! I think the diaminopyridine might have to be bought!
|
|
|
IPN
Hazard to Others
Posts: 156
Registered: 31-5-2003
Location: Finland
Member Is Offline
Mood: oxidized
|
|
I was originally going to make the picryl chloride from picric acid and phosphorus oxychloride as suggested by KABOOOM_[pyrojustforfun] but your
synth. starting from m-nitrobenzoic acid sounds much easier.
I will try it as soon as I get home.
|
|
|
IPN
Hazard to Others
Posts: 156
Registered: 31-5-2003
Location: Finland
Member Is Offline
Mood: oxidized
|
|
My attempt (half a year ago  ) at nitrating benzoic acid didn't go that well
(purifying the mixture was too tedious) and I forgot about making PYX. ) at nitrating benzoic acid didn't go that well
(purifying the mixture was too tedious) and I forgot about making PYX.
But as I was reading Vogel today I noticed the easy preparation of pure m-nitrobenzoic acid from methyl benzoate. Now this route seems much nicer.
Maybe in few weeks I'll be able to make the required picryl chloride.
EDIT:
Following the synthesis for methyl benzoate and m-nitrobenzoate in Vogel I ended up with milky oil floating in the crushed ice where the nitration mix
was poured. According to Vogel the nitrated ester should be a solid. What went wrong?
The methyl benzoate was distilled before the nitration and the nitration mixture was stirred for 18min in the ice bath after addition of the acids.
[Edited on 19.6.2005 by IPN]
|
|
|
IPN
Hazard to Others
Posts: 156
Registered: 31-5-2003
Location: Finland
Member Is Offline
Mood: oxidized
|
|
Here is a pic of the layers that formed.

|
|
|
Madandcrazy
Hazard to Others
Posts: 117
Registered: 11-5-2005
Member Is Offline
Mood: annoyed
|
|
Would the Chichibabin Reaction walking easy like in the diagram when refluxed with NaNH2 ?
Post not always the same things  in the forum. in the forum.
Maybe interested the sodium azide for this threath and the synthesis with the Cl-atom. Heterocyclic materials than furazanes or reacting around.
[Edited on 4-4-2006 by Madandcrazy]
|
|
|









