Aqua_Fortis_100%
Hazard to Others

Posts: 302
Registered: 24-12-2006
Location: Brazil
Member Is Offline
Mood: †
|
|
Reclaiming Pd from scrap SMD ceramic capacitors
Hello all.
Tried to search on forum (AKA UTFSE  ) but the only thing I found was a mention
of densest here: http://www.sciencemadness.org/talk/viewthread.php?tid=13225#... ) but the only thing I found was a mention
of densest here: http://www.sciencemadness.org/talk/viewthread.php?tid=13225#...
Few days back I was surfing through youtube and found occasionally an interesting video:
http://youtu.be/FWftjRnNrNw
Author in one of the comments claim:
| Quote: | Pd content vary with assorted MLCC's... it can be anywhere from 1%-2.5% by mass.
Also worth mentioning, is the silver content, in this case it was left behind as its chloride (AgCl) with the white powder.
Silver content can vary greatly, from 4.5% to 11% by mass.
|
His channel has other great experiments in precious metal recovering from e-scrap.
Picture I found at web showing the basic design of surface mount device (SMD) ceramic capacitor:
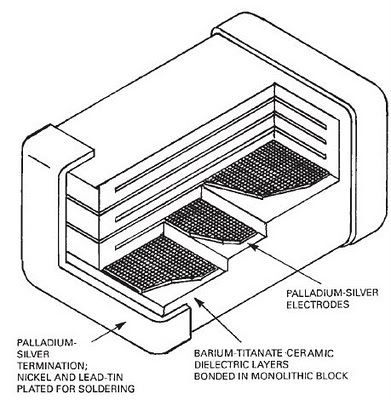
Found also a little thread on Gold Refining Forum about this (further refining process):
http://www.goldrefiningforum.com/phpBB3/viewtopic.php?f=51&a...
Since I have some scrap circuits containg these MLCC and lots of other things to recover/scavenge, I thought trying this would be interesting as Pd
have wonderful catalytic properties, can be used in anode making and of course is quite expensive.. Leaving silver, another interesting metal behind..
So lets start!
Here is all what Ive used:

Since I have very few electronic tools, I found myself that cuticle pliers of my mom did the job quite fast with ease.. (Although she wont be happy if
she know that  ). To remove it, just grab one of it and give a little torque, lift
and drop in a small bottle. ). To remove it, just grab one of it and give a little torque, lift
and drop in a small bottle.


Unfortunatelly, sometimes when you are in hurry (no need to say its a rather tedious process) you may apply to much force and/or grab the wrong way
and crush some of the MLCC and also sometimes terminations remain on circuit, so do the best to remove all of it.
Watch carefully all the way your scrap circuits before throw it as "done" as sometimes it can have MLCC and other SMD components on its back:

Here is again my small bottle of MLCCs (now with still more ):

This was from one motherboard and 4 small circuits.. But still dont weight it..
Cant wait to jump to next steps!
Have anyone made this before (or did I UTFSE wrong way  )? )?
Thanks!
[Edited on 13-1-2012 by Aqua_Fortis_100%]
"The secret of freedom lies in educating people, whereas the secret of tyranny is in keeping them ignorant."
|
|
|
Neil
National Hazard
Posts: 556
Registered: 19-3-2008
Member Is Offline
Mood: No Mood
|
|
Get a wood chisel or a cold chisel and push it along the board, you will depopulate the board in a few seconds.
|
|
|
Hexavalent
International Hazard
Posts: 1564
Registered: 29-12-2011
Location: Wales, UK
Member Is Offline
Mood: Pericyclic
|
|
Isn't there a chance you'll pick up resistors and what not with it as well though?
"Success is going from failure to failure without loss of enthusiasm." Winston Churchill
|
|
|
Neil
National Hazard
Posts: 556
Registered: 19-3-2008
Member Is Offline
Mood: No Mood
|
|
resistors contain Pt, just use a pair of optical sensors to plot the tools course.
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
A wood chisel! Excellent idea!
I've been using the hard (and smelly) road by intensely blowtorching the boards and then hitting them on concrete, where all the SMCs fly off in every
direction, along with the solder... ugh.
I have several large boxes filled with derelict PCBs that have been waiting to get "depopulated". Some parts are salvaged for my electronics hobby,
others are salvaged for precious metal reclaiming. I've been mostly saving PC card edges and CPUs, but would dearly love to salvage the platinum and
other rare metals found in all that electro-junk I have...
Robert
[Edited on 14-1-2012 by Arthur Dent]
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
Neil
National Hazard
Posts: 556
Registered: 19-3-2008
Member Is Offline
Mood: No Mood
|
|
You get bonus points if you attach a piece of carpet to a table and work on that. The fibers grip the pins like velcro and hold the board while you
work on it. You can easily use a sharp cold chisel with a hammer to strip pins with such a set up.
Nothing stinks like the magic smoke.
|
|
|
plante1999
International Hazard
Posts: 1936
Registered: 27-12-2010
Member Is Offline
Mood: Mad as a hatter
|
|
I have collected a small pile of smd , for refining silver and Pd, when I will see the results of aqua fortis I will attempt to do it.
I never asked for this.
|
|
|
Panache
International Hazard
Posts: 1290
Registered: 18-10-2007
Member Is Offline
Mood: Instead of being my deliverance, she had a resemblance to a Kat named Frankenstein
|
|
What? What? How old do they need to be?
|
|
|
Aqua_Fortis_100%
Hazard to Others
Posts: 302
Registered: 24-12-2006
Location: Brazil
Member Is Offline
Mood: †
|
|
Hello
Hi Neil, great idea.. I spend most time getting smd capacitors with cuticle pliers, wood chisel should be far faster!
Both resistors and capacitors contain precious metals, some times Pt, most times Pd (cheaper). Resistors have a thin layer of RuO2 too, maybe a kilo
of more of stuff should give useful recoverable amounts of ruthenium..

(Taken from: http://www.griederbauteile.ch/download/Datasheet/Widerstand/... ).
I get about 10g of MLCC and yesterday did a little test just to see..
About 0.4g was taken and put on test tube. Then about 2-3mL of 30% HCl was added.. Tiny bubbles instantly appeared.


Then I lit lighter to speed up things

Put a glass ball on tube's mouth to avoid excessive evaporation and let it stand for about 1 hour or so and then observed a tree-like crystall that
growed on solution-air interface (photo dont show very well):

I think this might be BaCl2*2H2O (leached from barium titanate dielectric).. Added a few drops of tap water (some cloudiness observed, the crystalls
re-dissolved then filtered..
To the filtered solution I added few drops of sodium sulfate solution, what I think proved to be barium:

Put in tube to decant (photo from partial decantation):

The residue of MLCC looks like about the same! The same weight (though my balance isnt very accurate)... So definately grounding it before leaching is
necessary (unlike the video).
I tested to magnetic residues (nickel, etc) some of the MLCC showed magnetic.
This is my magnetic separator.. Its only a syring piston with a neodymium magnet on end attached in an aluminium case. Just pull the piston and will
liberate/drop magnetic pieces out.


Will be making more tests today!
[Edited on 15-1-2012 by Aqua_Fortis_100%]
"The secret of freedom lies in educating people, whereas the secret of tyranny is in keeping them ignorant."
|
|
|
Aqua_Fortis_100%
Hazard to Others
Posts: 302
Registered: 24-12-2006
Location: Brazil
Member Is Offline
Mood: †
|
|
We posted almost the same time 
I heard of people that buy massive amounts of scrap/old USRR electronics for leaching, thats electronics are said to be one of the richest sources of
PMG.. Pt would be more present in these materials, as Urals Hills were(are) very rich in Pt.. So old USRR should gave it some use..
[Edited on 15-1-2012 by Aqua_Fortis_100%]
"The secret of freedom lies in educating people, whereas the secret of tyranny is in keeping them ignorant."
|
|
|
Aqua_Fortis_100%
Hazard to Others
Posts: 302
Registered: 24-12-2006
Location: Brazil
Member Is Offline
Mood: †
|
|
Hello.
More experimental data..
This time, weight 0.5g of MLCC, and crushed it using a mild steel frame of a disket/floppy and gentle beating with hammer.. I know ball milling should
be far better, but I dont have ball mill.


It wasnt perfectly crushed, but much better to react than before. So 20mL of HCl was added to a beaker containing this material and heated..

Heat was applied until about 10mL of liquid remained. SnCl2 test was negative:

It was left to cool somewhat and then a crust of crystalline precipitate was found above liquid and some at the bottom:


As previous experiments shows, this is supposed to be barium chloride.. Then about 30mL of tap water was added to dissolve it.. A interesting thing
happened.
Little barium titanate and other crap was found to be present this time, and as expected, water turned milky-cloudy..


But faster, the solution changed to light brown, dark brown, grey and finally black (air was present). I was thinking in iron presence, but black
colour is a bit puzzling to be iron.. Silver perhaps? Im decanting this solution to see..




Black (decanted in another container):

Then, after washing and decanting once more, it was time to play with Aqua Regia! ~2 mL of 30% H2O2 and 8mL of HCl 30% were put in the beaker with
MLCC residues and then, bits (~1mL at time) of 65% HNO3 were added with heating and with my nose far away..




Then, after most of NOx evolved, what remains was a somewhat red thick syrup

I could see white ppt (suposed AgCl). I added some mLs of demineralized water and a red ppt appeared..


This one is a crappy photo of SnCl2 positive test for Pd in this solution:

So this procedure will work.
Poor mans Pd/Pt/Ag ore!
Now, some questions:
What is the black powder?
The red powder is suposed to be Pd chloride, how could I re-dissolve it, excess HCl acid, it will form a complex?
Since I wasted few mLs of very precious (to me) HNO3 and I hate NOx, next time doing the procedure Im thinking in put some use for my homemade
chlorate.. (HCl/NaClO3 method).. what you think?
Thanks.
EDIT: It comes to my mind now: Instead of iron, could be the black ppt be some nickel compound?
[Edited on 15-1-2012 by Aqua_Fortis_100%]
"The secret of freedom lies in educating people, whereas the secret of tyranny is in keeping them ignorant."
|
|
|
Samuel-a
Harmless
Posts: 3
Registered: 15-1-2012
Member Is Offline
Mood: No Mood
|
|
Hi Aqua_Fortis_100%
Nice to see your experiment.
As i understand, the black solution was obtained by the second HCl addition, the orange [ish] color of the solution, before it turned black indicate
some Pd went in solution.
Probably the saturation of Ni,Sn,Pd and Ag form that color, it's not that unusual color.
AgCl (s) is at equilibrium with HCl solution.
Two main factors shift the balance toward the reactants side; Low temp' and dilution (H2O).
Decanting while hot, will take with it Ag+ ions, that will later ppt to form cloudy suspention as you witnessed.
This seems the case with you "red" powder ppt after Pd dissolution, AgCl pushed back out of solution, trapping some solution with in the crystalline
matrix, giving red/orange color.
I doubt it is PdCl2.
Cool with ice cubes the Pd solution for 30 minutes, and filter while cold.
Then reduce with Al/Zn and accumilate the ppt black powder, which now consist mostly of Pd. Don't expct much... probably 100-150 mg....
|
|
|
phlogiston
International Hazard
Posts: 1381
Registered: 26-4-2008
Location: Neon Thorium Erbium Lanthanum Neodymium Sulphur
Member Is Offline
Mood: pyrophoric
|
|
The white crystals you see forming in the HCl solution, as well as a the white precipitate may also very well be compounds of lead (from the solder).
Lead chloride is only sparingly soluble in cold water and much more so in hot water, consistent with the crystals you observe at the air/liquid
interfac in the heated HCl. The shape of the crystals and the way they from clusters of needles at the air/liquid interface are also similar to what I
observed upon recrystallising PbCl2 solutions.
Lead sulphate is very insoluble and will give a white precipitate upon adding any sulphate, like you observe.
Mixing solutions of soluble lead compounds (even PbCl2) with tap water will result in a milky, very fine precipitate in my experience, exactly like
you describe:
| Quote: | As previous experiments shows, this is supposed to be barium chloride.. Then about 30mL of tap water was added to dissolve it.. A interesting thing
happened.
Little barium titanate and other crap was found to be present this time, and as expected, water turned milky-cloudy.. |
I assume this is because of small amounts of sulphate, phosphate, carbonate ...? in tap water, which will give very insoluble products with lead.
So, for your own wellbeing, please note you may be dealing with very toxic solutions of lead compounds. (altough solutions of barium compounds are
pretty unhealthy as well).
-----
"If a rocket goes up, who cares where it comes down, that's not my concern said Wernher von Braun" - Tom Lehrer |
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
The way to do this is to melt all these ground MLCCs with either lead or silver (obviously with good ventilation) with a borax flux.. I had a client
that did these in a small furnace and slagged off the ceramic components in a borax flux but was getting quite a bit of palladium (I think he was
doing kilos of these per shot).
From there, you can digest it in 30% nitric acid, add sulfuric acid to precipitate out the lead as its sulfate, filter and then add HCl to precipitate
the silver from what should be a palladium solution. Filter out the AgCl, rinsing well with dilute HCl. Take that AgCl and cement it with nails and
sulfuric acid and re-use it for the next extraction. To the Pd aqua regia solution, concentrate that carefully on the steam bath until more AgCl or
other insoluble precipitate. Filter it again, then add a DMSO/EtOH solution of dimethylglyoxime to precipitate a voluminous yellow precipitate. This
can be ignited under the oxyhydrogen torch to Pd, or else decomposed carefully in a porcelain dish (very slowly to avoid PdO formation!).
Any Pt present will be left in the solution post DMG precipitation and can be recovered with zinc, boiled in sulfuric acid and then base, and then
re-digested in aqua regia when you've got enough to make it worth your while.
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
Arthur Dent
National Hazard
Posts: 553
Registered: 22-10-2010
Member Is Offline
Mood: entropic
|
|
Okay so to recap the procedure described above:
1) remove the chip capacitors and resistors from the board...
2) crush them with a hammer to make a coarse powder
3) put the powder in conc. HCl and heat the mixture until small purple-ish crystals form.
4) Add water and separate the soluble metal chlorides from the insoluble remaining debris
by decanting the solution from the solids.
The solution should contain mostly the soluble chlorides of Tin, Iron, Nickel, Barium, traces of Lead...
The solids may contain Palladium, Platinum and some Silver and Lead Chlorides
5) put the solids in HCl and add some Nitric Acid to produce Aqua Regia. Heat until all solids dissolve and produce an orange solution.
6) Test it with Tin Chloride for presence of Pt and Pd.
7) adding HCl to this solution should precipitate the silver first, leaving the Pt and Pd in solution.
8) Add metallic zinc, which will displace the Pd by precipitating it.
9) The final solution should contain Zinc and Platinum.
10) Test it with Tin Chloride for presence of Pt.
Is my account accurate, so far?
I'd be interested to try this during the weekend...
Robert
--- Art is making something out of nothing and selling it. - Frank Zappa ---
|
|
|
Samuel-a
Harmless
Posts: 3
Registered: 15-1-2012
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Arthur Dent  | Okay so to recap the procedure described above:
1) remove the chip capacitors and resistors from the board...
2) crush them with a hammer to make a coarse powder (The dust is very harmful, work with dust mask)
3) put the powder in conc. HCl and heat the mixture until small purple-ish crystals form.
4) Add water and separate the soluble metal chlorides from the insoluble remaining debris
by decanting the solution from the solids.
The solution should contain mostly the soluble chlorides of Tin, Iron, Nickel, Barium, traces of Lead...
The solids may contain Palladium, Platinum and some Silver and Lead Chlorides
5) put the solids in HCl and add some Nitric Acid to produce Aqua Regia. Heat until all solids dissolve and produce an orange solution.
6) Test it with Tin Chloride for presence of Pt and Pd.
7) adding HCl to this solution should precipitate the silver first, leaving the Pt and Pd in solution. (The solution is
already chloride based, more HCl will not change the solubility of Ag/Pb-Cl, adding some ice cube or ice bath or 3x times dilution is needed to remove
Ag/Pb-Cl from solution)
8) Add metallic zinc, which will displace the Pd by precipitating it. (Zinc will displace every metal beneath it on the
reactivity series, the black precipitate are subjected to following seperation and refining)
9) The final solution should contain Zinc and Platinum. (irelevant)
10) Test it with Tin Chloride for presence of Pt. (irelevant)
Is my account accurate, so far?
I'd be interested to try this during the weekend...
Robert
|
Good luck.
|
|
|
Aqua_Fortis_100%
Hazard to Others
Posts: 302
Registered: 24-12-2006
Location: Brazil
Member Is Offline
Mood: †
|
|
Thank you all people for the help!
| Quote: Originally posted by Samuel-a | Hi Aqua_Fortis_100%
Nice to see your experiment.
As i understand, the black solution was obtained by the second HCl addition, the orange [ish] color of the solution, before it turned black indicate
some Pd went in solution.
Probably the saturation of Ni,Sn,Pd and Ag form that color, it's not that unusual color.
|
Hello Samuel/IndeedItdoes, nice to see you sharing your experience on the video.
I doubt Pd went to solution after HCl attack, since SnCl2 test gaves negative result. Also, I did found in the moment that second portion of HCL was
not necessary as almost all ceramic stuff went to solution.. It was just dilluted with tap water.
| Quote: | AgCl (s) is at equilibrium with HCl solution.
Two main factors shift the balance toward the reactants side; Low temp' and dilution (H2O).
Decanting while hot, will take with it Ag+ ions, that will later ppt to form cloudy suspention as you witnessed.
This seems the case with you "red" powder ppt after Pd dissolution, AgCl pushed back out of solution, trapping some solution with in the crystalline
matrix, giving red/orange color.
I doubt it is PdCl2.
Cool with ice cubes the Pd solution for 30 minutes, and filter while cold. |
It has been long time since I read somewhere that AgCl forms complex with excess chloride, so when you told this I tried to search where I first saw
this.. and found it:
| Quote: | | It is well-known that silver chloride is highly insoluble, even in nitric acid; however, beginning chemistry texts often neglect to mention that
silver chloride will in fact form a soluble complex ion (AgCl2-) in the presence of excess chloride. AgCl is therefore soluble in excess NaCl or
concentrated HCl, especially when some AgNO3 is present (Merck Index, 1983). |
From: http://www.crscientific.com/article-silver.html (interesting to read in full! And has other nice experiments on minerals and related stuff)
Of course, I will be trying the ice-filter procedure next time! So, sad/angry that AgCl traped Pd 
| Quote: | | Then reduce with Al/Zn and accumilate the ppt black powder, which now consist mostly of Pd. Don't expct much... probably 100-150 mg....
|
Well, I made this anyway with residue/solution (without filtering it) yesterday, as I was very tired of my day. Will add that resulting material to
larger runs in future.. I was making these experiments with small amounts just to acquaint myself with the procedures and PMG refining before any
scale-up, as I didnt have any experience in playing with platinum metal group metals.. So now I have some to be proud! Though far from optimum for
decent metal recovering.
Added an little excess of fine homemade zinc powder:

This is a sequence of what followed:


After letting it react for about half an hour, I transferred it to a larger tube and dilluted (after do Pd test and have negative, of course). The ppt
was somewhat flocculent and some of it still with adhering bubbles, rised (acid on excess zinc). Swirled, decanted and filtered then.


| Quote: Originally posted by phlogiston | The white crystals you see forming in the HCl solution, as well as a the white precipitate may also very well be compounds of lead (from the solder).
|
Hello phlogiston.
Thank you! Have not thought on lead, but it may very well be present, as it has lower solubility than BaCl2! I have made lead chloride too (from PbCO3
and HCl.. And like silver, excess chloride will prevent considerable amounts of PbCl2 of ppting out as complexs forms -> PbCl3[-] and PbCl4[-2]).
Anyway, both barium and lead are poisons and very harmful to environment, so adding soluble sulfate salt to ppt both ions is a must to any home
chemist that doesnt want to have these soluble beasts floating around and get a chance to go back to you in worser ways..
But the 'bonus point' would be to separately ppt barium and lead and enrich your chemical shelves...And I have no clue on how to do this. Lead sulfide
is still more insoluble, but along with barium sulfide that is insoluble, is so nickel, etc.. With carbonate is the same story.
| Quote: Originally posted by Fleaker | The way to do this is to melt all these ground MLCCs with either lead or silver (obviously with good ventilation) with a borax flux.. I had a client
that did these in a small furnace and slagged off the ceramic components in a borax flux but was getting quite a bit of palladium (I think he was
doing kilos of these per shot).
|
Thats interesting, though I dont like to work with lead..Anyway should be a nice way to work with large amounts..
| Quote: | From there, you can digest it in 30% nitric acid, add sulfuric acid to precipitate out the lead as its sulfate, filter and then add HCl to precipitate
the silver from what should be a palladium solution.
|
What? Wouldnt you get fine Pd dispersion after 30% HNO3 leaching of Pd-Ag in lead matrix?
| Quote: | Filter out the AgCl, rinsing well with dilute HCl. Take that AgCl and cement it with nails and sulfuric acid and re-use it for the next extraction.
To the Pd aqua regia solution, concentrate that carefully on the steam bath until more AgCl or other insoluble precipitate. Filter it again, then add
a DMSO/EtOH solution of dimethylglyoxime to precipitate a voluminous yellow precipitate. This can be ignited under the oxyhydrogen torch to Pd, or
else decomposed carefully in a porcelain dish (very slowly to avoid PdO formation!).
Any Pt present will be left in the solution post DMG precipitation and can be recovered with zinc, boiled in sulfuric acid and then base, and then
re-digested in aqua regia when you've got enough to make it worth your while.
|
Unfortunately I dont have DMG, but have NH4Cl and Cl2, so the chloride/video process is better suited to me, at least for now..
Where goes nickel and tin?
| Quote: Originally posted by Arthur Dent | Okay so to recap the procedure described above:
1) remove the chip capacitors and resistors from the board...
2) crush them with a hammer to make a coarse powder
3) put the powder in conc. HCl and heat the mixture until small purple-ish crystals form.
4) Add water and separate the soluble metal chlorides from the insoluble remaining debris
by decanting the solution from the solids.
The solution should contain mostly the soluble chlorides of Tin, Iron, Nickel, Barium, traces of Lead...
The solids may contain Palladium, Platinum and some Silver and Lead Chlorides
5) put the solids in HCl and add some Nitric Acid to produce Aqua Regia. Heat until all solids dissolve and produce an orange solution.
6) Test it with Tin Chloride for presence of Pt and Pd.
7) adding HCl to this solution should precipitate the silver first, leaving the Pt and Pd in solution.
8) Add metallic zinc, which will displace the Pd by precipitating it.
9) The final solution should contain Zinc and Platinum.
10) Test it with Tin Chloride for presence of Pt.
Is my account accurate, so far?
I'd be interested to try this during the weekend...
Robert
|
As Samuel said, you will need to chill boiled aqua regia (what isnt no more AR) before filtering.
Of course larger amounts will make up for mechanical losses..
(Well, I may be forgetting something..)
"The secret of freedom lies in educating people, whereas the secret of tyranny is in keeping them ignorant."
|
|
|
Fleaker
International Hazard
Posts: 1252
Registered: 19-6-2005
Member Is Offline
Mood: nucleophilic
|
|
No, palladium is completely soluble in 30% nitric acid. I've dissolved Pd literally hundreds of times that way. Hell, you can even get platinum to
dissolve in nitric acid in some circumstances...
As for the chlorate and ammonium chloride method, check my post where I made a little bit of ammonium hexachloropalladate.
Neither flask nor beaker.
"Kid, you don't even know just what you don't know. "
--The Dark Lord Sauron
|
|
|
Samuel-a
Harmless
Posts: 3
Registered: 15-1-2012
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Fleaker | No, palladium is completely soluble in 30% nitric acid. I've dissolved Pd literally hundreds of times that way. Hell, you can even get platinum to
dissolve in nitric acid in some circumstances...
As for the chlorate and ammonium chloride method, check my post where I made a little bit of ammonium hexachloropalladate. |
Lou, is that you?
In few months i'll (hopefully) be involved in a contract for processing about 12-15 metric tons/year of capacitors.
sure would love to pick your barin about it when the time comes
|
|
|
Sedit
International Hazard
Posts: 1939
Registered: 23-11-2008
Member Is Offline
Mood: Manic Expressive
|
|
What are the odds of the product from this being useful to electroplate an electrode for performing a Kolbe synthesis?
How much of these would I possibly need to electroplate a 3x.5 inch electrode?
I have a ton of boards and have collected some of these but I don't want to process it till I have enough to be able to sufficiently coat an
electrode.
Knowledge is useless to useless people...
"I see a lot of patterns in our behavior as a nation that parallel a lot of other historical processes. The fall of Rome, the fall of Germany — the
fall of the ruling country, the people who think they can do whatever they want without anybody else's consent. I've seen this story
before."~Maynard James Keenan
|
|
|
Aqua_Fortis_100%
Hazard to Others
Posts: 302
Registered: 24-12-2006
Location: Brazil
Member Is Offline
Mood: †
|
|
| Quote: Originally posted by Fleaker | No, palladium is completely soluble in 30% nitric acid. I've dissolved Pd literally hundreds of times that way. Hell, you can even get platinum to
dissolve in nitric acid in some circumstances...
As for the chlorate and ammonium chloride method, check my post where I made a little bit of ammonium hexachloropalladate. |
You mean this thread: http://www.sciencemadness.org/talk/viewthread.php?tid=9721#p... ?
BTW, very nice thread! Thanks! Since I have formic acid and NH3 already, this would be an easier way to reduce Pd-Pt without worry about (NH4)2PdCl6
loss by sublimation!
Nice too Pd will dissolve that way (dilute HNO3), and since Ag will do also (quicker), theoretically after all in solution you only need to add the
right amount of HCl (very slight excess, not mean AgCl2[-]) to recover all your silver chloride before further Pd processing.
Since Im out my home and stay that way for a couple of days, I cant try right now this procedure.. But will do and compare with HCl-chlorate leaching.
| Quote: Originally posted by Sedit | What are the odds of the product from this being useful to electroplate an electrode for performing a Kolbe synthesis?
How much of these would I possibly need to electroplate a 3x.5 inch electrode?
I have a ton of boards and have collected some of these but I don't want to process it till I have enough to be able to sufficiently coat an
electrode. |
Amount its a matter of how thick the Pd film on electrode needs to be times total surface area.. And I dont know the former, but I guess you dont need
very much.. But thats just a guess, in that case start right now to collect your e-scrap Pd values and feedback we later with some pretty pics of your
process (since PMG chemistry is closely related to different coloured complexes and precipitates).
As stated earlier by experienced people, Pd content should be anywhere between 1 - 2.5% by mass.. So 100g of MLCC should give you 1 - 2.5g Pd, that
should suffice for small electrode in small amount of electrolyte..
EDIT: Oh crap, I forgot to mention I found a tiny treacherous crack on the bottom of the small beaker Ive used (the beaker shown couple posts above)..
I think Ive let it cool it too fast after AR treatment. The crack is only visible by swirling it against light (right angle with eye: invisible). So I
cant use it anymore with heating...My beaker is dead!
Will use a sand bath to heat other beaker next time and dont let it make contact with any room temperature/cool surface..
[Edited on 20-1-2012 by Aqua_Fortis_100%]
"The secret of freedom lies in educating people, whereas the secret of tyranny is in keeping them ignorant."
|
|
|
chemist1976
Harmless
Posts: 3
Registered: 2-4-2012
Member Is Offline
Mood: No Mood
|
|
Hi,
i'm now at the same procedure like aqua fortis on 15-1-2012 after heating in HCl.
I used 5.5 gramm of all smd (capacitors and resistors (Ruthenium)) and crushed them in a very hard work...!
After heating (1h at 90 degrees) the Solution of the "smd-powder" in 50ml 25%HCl the Solution gets a very nice violett color so what can it be?
The unsolutioble residue, not in the picture, looks grey/brown.
Could it be that in Solution is Ruthenium or/and Copper cause conc. HCl is able to dissolve Ru/Cu!?

|
|
|
RonPaul2012
Hazard to Self
Posts: 89
Registered: 29-2-2012
Member Is Offline
Mood: No Mood
|
|
I have my brother's old PS3 motherboard , does anybody know if it has any value (I mean for doing cool experiments with).
If so I will fish it out of the trash
[Edited on 8-4-2012 by RonPaul2012]
|
|
|
chemist1976
Harmless
Posts: 3
Registered: 2-4-2012
Member Is Offline
Mood: No Mood
|
|
first, excuse for my sometimes bad english, i wanna give my best.
| Quote: Originally posted by RonPaul2012 | | I have my brother's old PS3 motherboard , does anybody know if it has any value (I mean for doing cool experiments with). |
hmm, doing the self like me if you don't have better things to do - like me on sunday ;-) .
my question was only because i don't expect this colored solution.
ok, the "trash" i use contains Cu, Ni, Pb, Sn, Ag, Pd, Al, RuO and other you can find on every Mainboard.
second, before i use HNO3 with the residue, the next step, i wanna precipitate the elements i have in this solution.
i don't have learned the cation seperation at school and what i have in solution i can think by myself but i wanna "see" it.
is it better to boil the HCl away or neutralize it cause some Chlorids are volatile (Sn(IV)Cl), or not!?
|
|
|
lttadas
Harmless
Posts: 1
Registered: 15-1-2014
Location: Middle Europe
Member Is Offline
Mood: believing in tomorrow
|
|
Hi all,
i saw this topic read the whole thing and noticed that someone mentioned that Ag might be worthwhile recovering... So i done similar procedure as
Aqua_Fortis_100% . I took some electronic boards place them in a beaker with poor mans AR heated for some time after reaction was complete boards was
stripped clean only ceramic parts were left untouched. Then i decanted salution and left it to cool down to about 12 degress C. Next i filtered
precipitation it was white powder so i suspect it was Ag/Pb chlorides. Then i boiled white powder in distiled water and filtered white powder wich
turned out to became greyish after boilng. So my question would be if that greyish powder i got is pure AgCl or not ? Would appreciate quick response.
|
|
|









