| Pages:
1
2 |
Waffles SS
Fighter

Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
N-Methyl-d,l Alanine
As you know alanine prepare by reaction of a-bromopropionic acid with ammonia:
| Quote: |

Slowly and with stirring, 100 g. (0.65 mole, 59 cc.) of cold (1–4°) α-bromopropionic acid is added to 3 l. (44.5 moles, 2700 g.) of cold
(1–4°) concentrated aqueous ammonia (sp. gr. 0.9) in a 1-gal. glass-stoppered bottle, and the mixture is allowed to stand at room temperature for
at least four days . The solution is concentrated to a volume of 300 cc., filtered, and concentrated further to 200 cc. The solution is cooled to room
temperature and 1 l. of methyl alcohol added. After chilling overnight in a refrigerator (0–4°) the crystals are filtered with suction and washed
with 250 cc. each of methyl alcohol and ether. The yield is 42–48 g. of crude alanine.
For purification the crude product is dissolved in 200 cc. of water (warming if necessary), 1 l. of methyl alcohol is added, and the mixture chilled
overnight. After washing as before, the yield is about 38–42 g. (65–70 per cent of the theoretical amount) of purified dl-alanine, m. p. 295°
(dec.) on the Maquenne block (Note 7). This product is free of bromide and contains only traces of ammonia. If an especially pure product is desired
the material may be reprecipitated from methyl alcohol once more in the same manner.
http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv1...
|
and also this is possible to make N methyl d,l alanine by reaction of methyl amine (instead of ammonia) with a-bromopropionic acid.
But what about a- chloropropionic acid?(it is cheaper ,easier,safer to make)
| Quote: |
The use of α-chloropropionic acid gives a poorer yield (43–46 per cent of theoretical) and the product is more difficult to purify owing to the
fact that ammonium chloride is less soluble than the bromide in methyl alcohol. |
What is our problem in this reaction?(using a-chloro propionic acid)
Methylamine hydrochloride is more soluble in methyl alcohol than ammonium chloride
Dear cooker and amphetamine lovers,This topic isnt useful for you then please dont waste your time
[Edited on 23-4-2011 by Waffles SS]
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
I don't understand the extreme ratios, 3L of conc. nh3 (ick) to 100 grams?
Then 200 ml h20 and 1 liter of meoh for recrystallizing 42 grams?
It just seems wasteful as hell...
If you are really asking "what is the problem" its because Cl makes a stronger bond to carbon than bromide does, but I don't think that is what your
asking, is it?
I would assume slight warming would allow the equilibrium to shift slightly to the product, but as postart said you are going to get a mix of product
- not that you care of course.
An uneducated guess; if you raised the pH to 8 you may see more product...
|
|
|
mr.crow
National Hazard
Posts: 884
Registered: 9-9-2009
Location: Canada
Member Is Offline
Mood: 0xFF
|
|
Cl is a much worse leaving group than Br so it is less reactive. The huge excess of ammonia is so primary amines are favored. If it was less then it
would be more likely to produce secondary and tertiary amines.
Double, double toil and trouble; Fire burn, and caldron bubble
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
My question is about purify final product.
I think final product contain methyl amine hydrochlorodie,n-methyl d,l alanine, unreacted a-chloro propionic acid
and i am asking about n-methyl d,l alanine separation
In puryfing alanine ammonium chloride is less soluble than the bromide in methyl alcohol then this is difficult to purifing it if we use a chloro
propionic acid. but in the n -methyl alanine we have methyl amine hydochloride,
it is more solube in methyl alcohol than ammonium chloride and purifying should be better.isnt it?
|
|
|
postart
Hazard to Self
Posts: 59
Registered: 29-6-2010
Member Is Offline
Mood: No Mood
|
|
Why doesn't SWIM use fractional chromatography. N-methyl alanine is available commercially but may be looked at as an ephedrine precourser.
|
|
|
SovereignSolip
Harmless
Posts: 11
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
I don't really understand all of these thread looking for amphetamine precursors. All of the commercially viable routes are known and highly watched.
The ones that aren't watches, are not nearly as commercially viable. And if you really just wanted the stuff for personal use, there are quite a few
syntheses that are relatively cheap for a home chemist.
When these people get an inch, the try to take a whole mile.
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
leave drug-derailing out of the thread
I don't know why Waffles SS wants to make this substance, but if you search the American Chemical Society journals for n-methyl amino acid synthesis
it looks like a lot of other chemists are interested in the same topic. Amphetamines are simple enough molecules that, with a sufficiently jaundiced
eye, a lot of mundane chemicals look like amphetamine precursors. The first post in the thread explicitly says that the author is not looking for
advice on making controlled drugs. Don't spontaneously offer advice addressing the unasked and off-topic question "how can I easily make stimulant
drugs?"
PGP Key and corresponding e-mail address
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
that is one hell of an excess.
revealing the downsides of ammonolysis reactions the smaller the substrate molecule the larger the excess employed.
for example ammonolysis of methyliodide requires something like a 300 molar excess.
larger substrates that i've subjected to i could get by with 15 moles nh3.
i would suspect that would be suitable with a bulkier amine counterpart.
p.s. that equation is'nt balanced
[Edited on 7-5-2011 by jon]
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
This thread is full of so much bullshit. I don't care if you are making drugs or not; read more, post less.
Why is it hard to achieve good selectivity when alkylating amines? Generally as the number of alkyl substituents around the amine increases, so does
its nucleophilicity. Therefore, when alkylating an amine you have lots of unreacted product and a fair amount of poly-alkylated product and then not
much between.
There are many instances where amine alkylation is more stepwise; this is when alkylation produces an amine of lower nucleophilicity than the starting
material. Essentially, these are cases where the alkylating agent has some electron withdrawing groups on it. In these cases amine alkylation
becomes more stepwise.
The case of synthesis of a-amino acids is just this. Because the alpha-halo carboxylic acid has the electron withdrawing carboxylate group, it is
possible to have higher selectivity for alkylations, because the formed amino acids are generally weaker nucleophiles than the starting amines.
The classic synthesis of glycine from chloroacetic acid and ammonia is one such example.
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
no azo your not wrong it's just an impractical way to get alanine
and smuv good point there the formed amine would conjugate with the acid.
the only reason you would employ such an excess in a scheme like that is to prevent polyalkylation.
by surronding the substrate with ammonia molecules but since the product is rendered less reactive i don't see the point?
[Edited on 7-5-2011 by jon]
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
Some excess is still needed, N-methylaniline is only a little less nucleophilic than methylamine. That being said, I trust OS proceedures, but that
+50 fold excess of ammonia seems excessive to me (but then again, I am just handwaving). Water is not a great solvent to use the lower
nucleophilicity of amino acids to tune selectivity, something aprotic would be better. Also, the a-halo ester would probably allow for even greater
selectivity for mono-alkylation than the a-halo acid (which is largely deprotonated in solution).
"Titanium tetrachloride…You sly temptress." --Walter Bishop
|
|
|
jon
Hazard to Others
Posts: 459
Registered: 11-1-2006
Member Is Offline
Mood: paranoid distrustful apprehensive
|
|
doing ammonolysis on iodosafrole it only worked in isopropanol.
but it works really really well.
solvents play a big role in that reaction.
if methylamine were used you could use ethanol or methanol because it is a stronger nucleophile but still love ipa for those reactions
water kills those kind of reactions some water can be tolerated but not much more than 1%.
so it does'nt have to be bone dry.
[Edited on 8-5-2011 by jon]
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
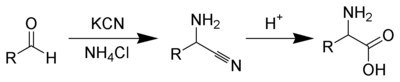
I think this is possible to make N-Methyl-d,l Alanine by Strecker amino-acid method
http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv1...
Am i wrong?
|
|
|
Jesse Pinkman
Harmless
Posts: 13
Registered: 28-8-2012
Member Is Offline
Mood: No Mood
|
|
I have one idea:
1) Oxidation of lactic acid to the corresponding carbonyl compound
2) Reductive amination of the carbonyl compound to the desired amine

|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
somebody tried tetrahedron letter on N-methylation L-alanine by formaldehyde + Zinc?
I Like to test this method but i dont have detail for this reaction,this letter has little info about reaction detail
Attachment: Zn_HCHO_methylation.pdf (98kB)
This file has been downloaded 2439 times
|
|
|
Dr.Bob
International Hazard
Posts: 2734
Registered: 26-1-2011
Location: USA - NC
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Waffles SS  | As you know alanine prepare by reaction of a-bromopropionic acid with ammonia:
and also this is possible to make N methyl d,l alanine by reaction of methyl amine (instead of ammonia) with a-bromopropionic acid.
But what about a- chloropropionic acid?(it is cheaper ,easier,safer to make) |
The chloro group in -chloropropionic acid is just not as good of a leaving group as the bromo is in the a-bromopropionic acid. So it will react
slower. That may help reduce di or tri aklylation of the nitrogen, but will also slow down reaction and lower yield, most of the time.
As for the large excesses used, this is an older prep, where they relied on crystallization to collect the product, so driving the reaction as far as
possible with cheap reagents was normally done then. Also, it was harder to monitor reactions for completion, so again, going overboard was common.
If you just want the N-Me-(D,L)-alanine, this should work OK with N-methylamine, but I would expect that there are better ways to make the compound,
especially if you want the chiral material. I would make the formamide of alanine and reduce it, as that can be done without racemizing, I believe.
The Tett Letters paper is short on details, but they claim that running the reaction only 15 minutes generates the monoalkylated alanine in 85% yield.
Not sure how you would purify that from the diMe easily without going further steps. It sounds pretty easy to test, and they give some basic
conditions in their table.
[Edited on 26-9-2012 by Dr.Bob]
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
Thanks Dr.Bob,
I tried this reaction by below ratio:
28 gr Sodium Di Hydrogen Phosphate ,12gr Formalin(37%),15gr Zinc dust,10gr L-alanine(i just have access to L-alanine) and 200ml water.
After adding all of these, tiny bubbles start to come(Hydrogen) and after 15 minutes you can see white powder on surface of Zinc dust(Zinc Phosphate).
I dont know how can i separate N-methyl from solution.suggested solvent by letter(Chloroform ) didnt work because N-Methyl is soluble in water too.
I think another possible route for making N-methyl is also possible
First : Preparing 2-chloropropionic acid from L-alanine via diazotization in hydrochloric acid
(http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv8...)
Second : Preparing alanine by ammonolysis of 2-bromopropanoic acid(also N-Methyl-alanine by Methylammonolysis of 2-chloropropanoic is possible)
(http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv1...)
this article use bromopropionic acid and ammonia but as you know and mentioned on article ,ammonolysis of chloropropionic acid is possible(and also
Methylammonolysis)
[Edited on 27-9-2012 by Waffles SS]
|
|
|
Rich_Insane
Hazard to Others
Posts: 371
Registered: 24-4-2009
Location: Portland, Oregon
Member Is Offline
Mood: alive
|
|
| Quote: |
I have one idea: 1) Oxidation of lactic acid to the corresponding carbonyl compound 2) Reductive amination of the carbonyl compound to the desired
amine |
I like this idea, but is it also possible to brominate lactic acid by reaction with HBr? I understand that in situ generation via
H2SO4/Alkali bromide would create competition for the dehydration, but will lactic acid even brominate efficiently with HBr?
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
I am confused.
I think L-alanine should be L(+)Alanine,(+)dextrorotatory,(s)eniantomer and Optical Rotation is +13.5° ~ +15.5° but in orgsyn mentioned (S)-Alanine
Optical Rotation is −13.98 !!
http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv8...
Which one is correct?For making 2-ChloroPropionicAcid by diazotization and react it with Methylamine for making N-methyl-L-alanine Which alanine
should be used?
|
|
|
Rich_Insane
Hazard to Others
Posts: 371
Registered: 24-4-2009
Location: Portland, Oregon
Member Is Offline
Mood: alive
|
|
Wouldn't it be best just to use racemic alanine?
|
|
|
Salmo
Harmless
Posts: 42
Registered: 20-9-2012
Member Is Offline
Mood: No Mood
|
|
Man I think you have to check the diazotization stereoselectivity, I think that diazotization shouldn't cause inversion of the chiral center, but i
could be really wrong and I didnt check about the stereoselectivity about the reaction with methylamine even if I think that doesen't cause inversion
too.. anyway I found this pdf for you, read pag 87.
Attachment: FULLTEXT01.pdf (1.8MB)
This file has been downloaded 2133 times
[Edited on 1-10-2012 by Salmo]
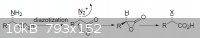
[Edited on 1-10-2012 by Salmo]
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
Thanks salmo for your helpful text.according to this text diazotization dont cause inversion of the chiral center .this mean that diazotization of
L-alanine cause R(+)2-ChloroPropionicAcid(am i wrong?)
I really dont know what Orgsyn wrote(Optical Rotation of (s)alanine is +13.5 not -13.5)
L-alanine(Synonyms : (S)-Alanine,L-(+)-Alanine,(S)-(+)-Alanine) OPTICAL ROTATION +13.5° ~ +15.5°
R(+)2-ChloroPropionicAcid OPTICAL ROTATION +14°
[Edited on 2-10-2012 by Waffles SS]
|
|
|
aliced25
Hazard to Others
Posts: 262
Registered: 31-7-2010
Member Is Offline
Mood: No Mood
|
|
It can also be prepared by the reductive amination of pyruvate esters with the appropriate amine. I suspect other reductive approaches would work just
as well.
I do wonder though, could that imine, which is very similar in structure to the one formed by the reaction of benzaldehyde with alanine esters, be
alkylated directly at the 2-carbon giving a-methylphenylalanine directly (with reductive n-debenzylation).
Would be a short route to a-methylphenylalanine if it could
Attachment: Crouch.Holden.Weaver.Reductive.Amination.of.Pyruvate.Esters.A.Microscale.Synthesis.of.N.Benzylalanine.Esters.pdf (53kB)
This file has been downloaded 885 times
From a Knight of the Realm: "Animated movies are not just for kids, they're also for adults who do a lot of drugs." Sir Paul McCartney
|
|
|
phlogiston
International Hazard
Posts: 1379
Registered: 26-4-2008
Location: Neon Thorium Erbium Lanthanum Neodymium Sulphur
Member Is Offline
Mood: pyrophoric
|
|
| Quote: | 28 gr Sodium Di Hydrogen Phosphate ,12gr Formalin(37%),15gr Zinc dust,10gr L-alanine(i just have access to L-alanine) and 200ml water.
|
You don't say anywhere if you desire a racemic mixture or not, but if you do I found an easy method to quickly obtain a racemic mixture of most amino
acids is to heat them with pyridoxal-5-phosphate (vitamine B6) in an alkaline environment.
I only occasionally need small amounts for analytical purposes. Last time I took about 40 ul of a 25 mM solution of L-amino acid + 250 ul 25% ammonia
+ 10 ul 100 mM PLP. This was heated in a glass vial with a screw cap to 95 deg C and kept at this temperature for 1 hour, resulting in complete
racemisation and 90% recovery.
If you need to get rid of the PLP and its products you can use W50 cation exchange resin.
It works well for most but not all amino acids.
The following fragment (from wikipedia) may clear up the confusion regarding the optical rotation:
| Quote: | The D/L labeling is unrelated to (+)/(− ; it does not indicate which
enantiomer is dextrorotatory and which is levorotatory. Rather, it says that the compound's stereochemistry is related to that of the dextrorotatory
or levorotatory enantiomer of glyceraldehyde—the dextrorotatory isomer of glyceraldehyde is, in fact, the D- isomer. Nine of the nineteen L-amino
acids commonly found in proteins are dextrorotatory (at a wavelength of 589 nm) ; it does not indicate which
enantiomer is dextrorotatory and which is levorotatory. Rather, it says that the compound's stereochemistry is related to that of the dextrorotatory
or levorotatory enantiomer of glyceraldehyde—the dextrorotatory isomer of glyceraldehyde is, in fact, the D- isomer. Nine of the nineteen L-amino
acids commonly found in proteins are dextrorotatory (at a wavelength of 589 nm) |
-----
"If a rocket goes up, who cares where it comes down, that's not my concern said Wernher von Braun" - Tom Lehrer |
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
| Quote: Originally posted by phlogiston |
You don't say anywhere if you desire a racemic mixture or not, but if you do I found an easy method to quickly obtain a racemic mixture of most amino
acids is to heat them with pyridoxal-5-phosphate (vitamine B6) in an alkaline environment.
I only occasionally need small amounts for analytical purposes. Last time I took about 40 ul of a 25 mM solution of L-amino acid + 250 ul 25% ammonia
+ 10 ul 100 mM PLP. This was heated in a glass vial with a screw cap to 95 deg C and kept at this temperature for 1 hour, resulting in complete
racemisation and 90% recovery.
If you need to get rid of the PLP and its products you can use W50 cation exchange resin.
It works well for most but not all amino acids.
|
Thanks @phlogiston,
Can you put reference for this method?
No different.
Somebody ever tried separation of amino acid by Isoelectric Point?I tried L-alanine and glycine but i failed.
I dissolved 10 gram L-alanine in 100cc water and set PH to isoelectric point of alanine and then cool solution to 4-5c for 24 hours but no crystal
appeared.!
Somebody ever tried to resolution racemic mixture by Chiral Column Chromatography?
[Edited on 6-6-2013 by Waffles SS]
|
|
|
| Pages:
1
2 |