| Pages:
1
2 |
Monoamine
Hazard to Others

Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Project: Making thionyl chloride from easy-to-get precursors
Warning: SO2, HCl and Cl2 are toxic gasses. SO3, SCl2 and SOCl2 are
all extremely corrosive and react violently with water.
This is a set of recipes/notes on how to synthesis thionyl chloride (SOCl2) from easy to get materials. The project will be organized into
a seperate post for each separate step/experiment all within this thread. Participation and discussion is welcomed.
Ever since I learned about it, the reagent thionyl chloride SOCl2 has fascinated me. It’s very useful as a chlorinating agent, can dry
many reagents and is very “clean” to use, in the sense that when it reacts, the byproducts leave the reaction as gases. That being said, it’s
also somewhat hazardous because said gases are sulfur dioxide SO2 and hydrogen chloride (HCl), which is why this reagent is particularly
hard to get for the amateur scientist.
Therefore there exists a need for a home-synthesis of thionyl chloride which uses precursors that should be in the reach of most folks as well as
being relatively inexpensive. Fortunately, the need for expensive, hard to get equipment and chemicals can be circumvented by anyone with patience and
an honest interest in science. In this thread we will carry out a series of experiments aimed at showing that this is indeed possible and how it can
be done. The hardest to get chemical in the entire synthesis is probably concentrated H2SO4 and - as we shall see - only a
catalytic amount is needed.
SOCl2 is formed when sulfur trioxide (SO3) reacts with sulfur dichloride (SCl2). Since we are making everything from
easy to get chemicals, the SOCl2 is synthesized in three steps:
Step 1: Synthesis of SO3
SO3 is produced by heating Na2S2O8 with a small amount of catalytic H2SO4 to 300C.
(Without the catalyst, the temperature required is over 460C).
2Na2S2O8 -> 2Na2S2O7 + O2
Na2S2O7 -> Na2SO4 + SO3
Step 2: Synthesis of SCl2:
SCl2 is produced by generating Cl2, passing it over elemental sulfur and distilling of SCl2 as it forms.
Ca(OCl)2 + 4HCl -> CaCl2 + 2H2O + 2Cl2 (Your method of Cl2 generation may differ).
2S + Cl2 -> S2Cl2
S2Cl2 + Cl2 <-> 2SCl2
Step 3: Synthesis of SOCl2 from SO3 and SCl2:
SO3 + SCl2 -> SOCl2 + SO2
Ingredients and where to get them (the amounts are only approximate):
200ml concentrated sulfuric acid (H2SO4). Can usually be ordered online or distilled from some brands of drain cleaner. Note
that only 30ml-50ml are actually used up in an ideal synthesis, the rest can be reused.
2kg Sodium persulfate (Na2S2O8). Used in etching copper and electronic circuits. Look in stores where they sell
etching supplies.
150g elemental sulfur (S). Sold in some pharmacies and online.
1.5kg calcium hypochlorite Ca(OCl2). Used as a swimming pool chlorinating agent. 2-3L
conc. hydrochloric acid (HCl). Sold as muriatic acid in swimming pool supply stores.
Essential equipment:
Distillation setup.
Safety equipment: Gloves and eye protection. Gas mask with military grade filter, or fume hood.
Reflux condenser.
Two necked flask.
Hoses.
Heating mantle or hotplate capable of reaching at least 300C.
Heatgun.
Glassware or other material needed to make chlorine.
Recommended equipment:
Drying tube.
Glass funnel.
NaOH for apparatus drying and quenching of acidic fumes.
Full face protection.
Protective overcoat.
Cl2 generator (if Cl2 is made in-house):
Gas wash bottle.
Pressure equalizing addition funnel.
Suckback trap.

Let the fun begin!
[Edited on 14-12-2021 by Monoamine]
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Synthesis of sulfur trioxide (SO3)
This procedure is adapted from the procedure described by user “Garage Chemist”. His original notes can be found here.
In brief: Sodium pyrosulfate (Na2S2O7) can be formed by heating sodium persulfate
(Na2S2O8). Na2S2O7 decomposes above 460C and gives SO3. The temperature
required for this decomposition can be lowered to around 300C by employing a catalytic amount of H2SO4.
Previously we attempted this synthesis with a few mishaps. Consult this post for a discussion of what can go wrong and potential pitfalls.
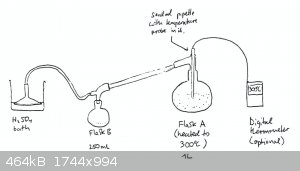
To a 1L round bottom flask (flask A) was added 800g of sodium persulfate (Na2S2O8). To this was added some broken
glass pieces as boiling chips and no stir bar, since the SO3 can actually dissolve the stir bar (learned this during the first trial…).
Finally, 15ml of 95% H2SO4 was also added to flask A.
This was then set up for simple distillation. But the condenser was NOT cooled with water. The joints of the setup were “greased” with 95%
H2SO4, by using an old nitrile glove dipped in H2SO4 to apply it. This is highly recommended and greatly
helps to reduce fumes escaping the apparatus. Flask A was placed into a heating mantle and wrapped in aluminum foil. It was found that not wrapping
the flask and still head in aluminum foil makes the process agonizingly slow.
The vacuum takeoff adaptor was connected to an inverted funnel over a 95% H2SO4 bath in a beaker in order to dissolve
SO3 fumes and protect the apparatus from moisture. One neat feature of this is moisture trap is that any SO3 that is dissolved
in the H2SO4 is converted to H2SO4, so when you later go to retrieve the H2SO4 in
the beaker, you’ll probably have purer H2SO4 then you started with!
Flask B was filled with about 50ml of 95% H2SO4 to give the SO3 something to dissolve into. In retrospect, this is
probably not too great an idea, because it decreases the yield because the SO3 first has to dehydrate the H2SO4.
A thermometer probe that was housed in a glass pipette whose tip had been melted shut was inserted into flask A. The SO3 will attack any
thermometer not made of glass, and non-digital thermometers are usually not able to read temperature in the 300C-350C range, needed here. That being
said, using a thermometer isn’t really necessary for this synthesis. Turning the heating mantle to maximum and wrapping the flask in aluminum foil
held the temperature at around 320C in the still head. This will however also distill off your catalytic H2SO4. The catalytic
H2SO4 will do its job before distilling away.

The heating mantle was turned to maximum.
At first, at around 100C, a lot of bubbling was observed in the H2SO4 bath. This is from evolving O2 via the
reaction:
2Na2S2O8 -> 2Na2S2O7 + O2
As the temperature in the still head rises to 300C the contents of flask A melts and is converted to SO3 and Na2SO4
via the reaction:
Na2S2O7 -> Na2SO4 + SO3
At this point vapours filled the apparatus and small clear drops of SO3 began to come over. Some vapour even came out of the upturned
funnel in the H2SO4 bath. There was also some brown discolouration in the SO3 that comes over which is due to the
SO3 oxidizing residual organic matter in the glassware.
Occasionally there was a build up of solid SO3 in the apparatus. If this clogs the setup it can be very dangerous. It never got to that
point in this run, but even so, a heat gun was used to warm the glass around frozen SO3 to melt it into flask B.
After about 3-4 hours the drip rate had become vanishingly slow and the distillation was stopped.
Flask B was stoppered and stored in a, glass jar with a lid, on a bed of dry Na2CO3.

The apparatus was not washed with water to avoid the glass from shattering due to residual SO3 reacting with H2O and instead was
simply left outside under an umbrella to allow the residual SO3 to slowly react with moisture in the air overnight. It was then properly
rinsed out the following morning.
Notes:
Using a simple glass column rather than a condenser is recommended, since it makes heating the column to melt solid SO3 much easier, and
water cooling was never needed.
I only noticed the need for greasing the joints with H2SO4 when the distillation had already started, but needless to say, this
should be done before hand.
Unless you want to store the SO4 very long term, I would recommend against putting any H2SO4 in the receiving flask,
since it decreases the yield quite a bit. (In fact, I think if your aim is making SOCl2, then just distilling the SO3 directly
into SCl2 is probably the way to go).
If the synthesis of SCl2 was performed before this synthesis, then it’s probably a good idea to use the H2SO4 used
for drying the Cl2 in the acid bath under the upturned funnel. That way the more wet H2SO4 from the Cl2 drying can
itself be dried by escaping SO3 and then recycled for later batches. This way the only H2SO4 that is ever used up in this
synthesis is the catalytic H2SO4 used to make SO3.
Something that is very strange in all of this is that the content of flask A is a liquid even at temperatures as low as 250C and when almost nothing
more distills over. At this point the only thing that should still be in the flask is Na2SO4 and maybe unreacted
Na2S2O7 and a very small amount of H2SO4. Considering that there is only a very small amount of
sulphuric acid present and the melting points of Na2S2O7 and Na2SO4 are all above 400C, this
does not make any sense. The content of the flask should be solid… Any ideas why it isn’t?
This procedure also seems to be a relatively easy way to make 100% sulfuric acid.
[Edited on 14-12-2021 by Monoamine]
[Edited on 15-12-2021 by Monoamine]
|
|
|
SWIM
National Hazard
Posts: 970
Registered: 3-9-2017
Member Is Offline
|
|
I had no idea the pyrosulfate would decompose at such a low temperature with a little added H2SO4.
This looks a lot simpler and less destructive to equipment than the dehydration by metaphosphoric acid.
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Synthesis of sulfur dichloride (SCl2)
This synthesis is adapted from a youtube video by Extraction&Ire (found here). Also consult the wiki for this synthesis.
This is the synthesis which was maybe the easiest to carry out and produced the largest amount of product. It’s also the step which requires the
most pieces of apparatus, but much of it can be home built. (I made two of the gas wash bottles out of rubber stoppers and glass straws and the
pressure equalizing addition funnel out of a separatory funnel, a Claisen adapter and some hoses).
The reaction proceeds by passing Cl2 into sulfur and distilling off the SCl2 that forms.
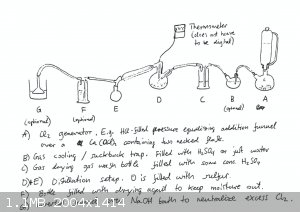
To generate chlorine (Cl2) gas, a Cl2 generator was put together as follows: To a separate two necked 500ml round bottom flask
(flask A) was added about 100g of calcium hypochlorite (Ca(OCl)2), and to this was connected a 250ml (very clumsily improvised) 250ml
pressure equalizing addition funnel that was filled with 32% hydrochloric acid.

The two necked flask was then connected to a suckback trap (flask B) with a hose, since the Ca(OCl)2 has a risk of foaming over when
reacting with HCl. For this reason it is also recommended to use at least a 1L two necked flask to house the Ca(OCl)2, but I just don’t
happen to have one. The suckback trap was then connected to a gas wash bottle (bottle C) containing about 50ml of 95% H2SO4, for
gas drying.
Bottle C was then connected with a hose to a 500ml round bottom flask (flask D) in a heating mantle. Flask D was filled with 170g of elemental sulfur
and a magnetic stir bar. Flask D was then connected to a water-cooled Liebig condenser and a thermometer was inserted into flask D.
Flask E is used to collect our SCl2, and the gas takeoff adaptor was connected with a hose to a (again very makeshift) bottle containing
dry Na2CO3 which serves to form a moisture barrier and can also neutralize some of the excess Cl2 that escapes.
Finally, Flask E was connected with a hose to an upturned funnel in a NaOH bath to further neutralize Cl2 and any other acidic fumes that
are produced in the process.

SCl2 was then produced as follows:
1) HCl from the addition funnel above flask A is dripped onto the Ca(OCl)2 in flask A. This generates Cl2 via the reaction:
Ca(OCl)2 + 4HCl -> CaCl2 + 2H2O + 2Cl2
2) This generates a pressure gradient towards the “exit” at F, and the Cl2 is passed through the H2SO4 in bottle
C, which strips it of moisture and dries it.
3) The dry Cl2 is then passed into flask D where it reacts with the sulfur to form disulfur dichlorideS2Cl2, and –
if an excess of Cl2 is available – sulfur dichloride SCl2. This happens in two steps via the reactions:
2S + Cl2 -> S2Cl2
S2Cl2 + Cl2 <-> 2SCl2

4) Note that the formation of SCl2 is an equilibrium reaction and that it slowly decomposes back into chlorine and disulfur dichloride,
especially if sulfur is still present.
5) Luckily the boiling point of SCl2 is 59.6C and that of S2Cl2 is 138C, so SCl2. Therefore, we distill
off the SCl2 as it forms and collect it in flask E.
6) In the end, this produced about 200ml of SCl2.
This was carried out, and we re-filled the flask A with fresh Ca(OCl)2 whenever no more Cl2 was generated, until no more
SCl2 distilled over. All in all, we used about 800g of Ca(OCl)2 and 2L of 32% HCl.
We found that adding only a small amount of Ca(OCl)2 to the Cl2 generator greatly increases the efficiency of the transformation
to Cl2 and also prevents overfoaming. Unreacted Ca(OCl)2 can be made to react with HCl in the flask by gently shaking the flask.
Despite trying to quench much of the excess Cl2 that was made, a great deal still escaped and I stank like a swimming pool for a day. The
Cl2 also completely oxidized the connecting hoses as well as the metal lab stands and Keck clips. So just to re-emphasise, a good way to
protect yourself from toxic gasses is essential here.
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Discussion: Sulfur dichloride

SCl2 slowly decomposes back into S2Cl2 and Cl2. For this reason, we did not want to seal it into a
completely tight container to avoid pressure build up. On the other hand, SCl2 also react violently with water and fumes as soon as it gets
in contact with the moisture in the air. As a compromise, we put it into a glass bottle with a glass stopper and only gently stoppered it, so that
pressure could still be released. The bottle was then put into an open beaker on a bed of Na2 CO3. This way, if Cl2
escapes the bottle it will stay in the beaker and be neutralized by the Na2CO3 (since Cl2 is heavier than air). We
have now stored the SCl2 for a week in this fashion and have not had any problems.
Question: I couldn’t find a clear answer as to how SCl2 reacts with alcohols and carboxylic acids. Does anyone know if
it can act as a chlorinating agent in of itself already?
Since this reaction involves a lot of acidic fumes, we surrounded the apparatus with a few concentric rings of litmus paper. This way, as acidic fumes
escaped the apparatus, the distance that they travelled and the amount could be visually gauged.

Above you can see that the litmus paper closer to the apparatus is already quite red.
Great! Now we have all the precursors to make thionyl chloride.
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Very nice work! Especially the SO3 generation -- that's a much more convenient setup than any other I've heard described.
| Quote: | | I couldn’t find a clear answer as to how SCl2 reacts with alcohols and carboxylic acids. |
If it seems obvious that two chemicals react, but nobody describes the reaction, there's a good chance the reaction produces a "complex mixture".
Not stoichiometric:
SCl2 <> S2Cl2 + Cl2
S2Cl2 + ROH >> RCl + S2O (yes this is a real compound)
S2O + Cl2 >> SOCl2 + SCl2 etc
S2Cl2 is more stable and less likely to oxidatively chlorinate the substrate, which I think is why it is preferred for deshydroxy-chlorination
reactions.
[Edited on 15-12-2021 by clearly_not_atara]
|
|
|
woelen
Super Administrator
Posts: 8012
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Very nice result, both the making of SO3 and the making of SCl2!
Heating a glass vessel to well over 300 C for making SO3 still is quite scary, but indeed, this is much more doable than heating dry Na2S2O7 for
getting SO3 and can be done with (good quality and reliable) glassware.
Thumbs up!
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Thank you clearly_not_atara for the tips about reactions of SCl2 with alcohols. Maybe a future project might be trying to see if it can be used to
make alkyl chlorides.
|
|
|
DraconicAcid
International Hazard
Posts: 4332
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Online
Mood: Semi-victorious.
|
|
I'm very interested to learn if SCl2 could be used to convert an acid into an acid chloride....
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
Lionel Spanner
Hazard to Others
Posts: 168
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Quote: Originally posted by Monoamine  | | Question: I couldn’t find a clear answer as to how SCl2 reacts with alcohols and carboxylic acids. Does anyone know if
it can act as a chlorinating agent in of itself already? |
It mostly seems to be used for making chlorine-substituted thioethers from alkenes, S/N heterocycles from nitriles, and various polymeric species.
There needs to be a higher oxidation state at sulphur (IV or VI) to transfer chlorine without transferring the sulphur as well.
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Thionyl chloride synthesis (SOCl2) from sulfur trioxide (SO3) and sulfur dichloride (SCl2). Phase I.
To make SOCl2, we followed the synthesis of Georg Brauer on page 382 in “handbook of preparative inorganic chemistry”. A pdf of this
wonderful book can be found here.
In brief:
SO3 is distilled into SCl2, where it reacts and forms SOCl2 and SO2 via the reaction:
SO3 + SCl2 -> SOCl2 + SO2.
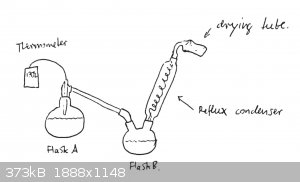
A round bottom 250ml flask (flask A) containing all of our oleum (from experiment 1 and some earlier trials) was set up for distillation. As the
receiving flask, we used a 500ml two necked round bottom flask (flask B) connected to a Graham condenser that was fitted with a drying tube containing
NaOH. Flask B was filled with all of our SCl2 (a little over 250ml). (In retrospect, far less SCl2 should have been used.)
Neither the Graham condenser nor the Liebig condenser were cooled with water. Feeling them periodically, however showed that they were always cold to
the touch.

Flask A was heated slowly. At about 135C, a white vapour started blowing into flask B. The formation a kind of light curst in flask B was observed.
At about 150C the first drops of liquid started collecting in the condenser. At this point I realized that I should have greased the joints of the
distillery with H2SO4 before starting the experiment, because some fumes started to leak. So I quickly pulled the apparatus open
and applied some panicky sulfuric acid to the joints to seal them. This worked to stop the fumes from escaping, but I probably also lost some
SO3 in the process, because every time the apparatus is opened, thick white vapours billow out (this is not recommended).
At 175C, a quick and steady drip rate had been achieved. Yellowish solidification was observed in flask B and the temperature in flask B rose to
being `warm to the touch’ and some gentle refluxing occurred in flask B.
The reaction seems to be locally exothermic. As soon as a drop of SO3 hits the SCl2, it fizzles and a small amount of solid
forms (not sure why solid forms??)
When the distillation was done, there was a great deal of yellowish solid in the red liquid. Using a heatgun, the solid could easily be melted into
the SCl2, where it seemed to react with the SCl2.

The melting point of the solid seems quite low, so maybe some of this is sublimed SO3? On the other hand, this does not explain why there
are also chunks of solid submerged in the SCl2?? That being said, Brauer also comments that solidification of the consent of flask B is
frequently observed in the beginning, but he does not say why this happens or what it is.
Also, at this point the whole shed I was working in completely reeked of fireworks and rotten eggs, which I think is the smell of sulfur dioxide?

Once all of the yellow solid had been melted into the solution in flask B, the colour turned to a darker red and a chunk of solid remained at the
bottom of flask B.
In phase two we attempted to separate the SCl2 and SOCl2 in flask B by fractional distillation.
References:
Brauer G, editor. Handbook of Preparative Inorganic Chemistry V2. Elsevier; 2012 Dec 2.
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Thionyl chloride synthesis (SOCl2) from sulfur trioxide (SO3) and sulfur dichloride (SCl2). Phase II.
Next, we stoppered flask B and set it up for distillation to try and fractionally distill off the SCl2 and any SOCl2 it
contained.

At 35C, a significant amount of fumes smelling like rotten eggs and fireworks (SO2?) evolved from the drying tube above the collection
flask. Holding a solution of 25% ammonia close to the opening of the drying tube showed that these fumes are quite acidic as the ammonia neutralized
them and thick grey vapour was created.

The solid in the SCl2 + SOCl2 flask also disappeared, so maybe there was somehow SO3 locked up in the solid in the
flask, which then melted into the solution and reacted with SCl2 to make more SOCl2?
The boiling point of SCl2 is 59.6C and the boiling point of SOCl2 is 74.6C.
Orange distillate started coming over at 50C already (which is 9.6C lower than the boiling point of SCl2), but judging by the colour, it
surely must be SCl2.

The temperature was increased to above 65C and then varied between 73C and 57C while the orange droplets of SCl2 continued to come over.
A lot of yellowish solid began to build up near the end of the condenser. I’m actually not sure what this is but it seems to be due to a process in
the SCl2 alone since the same build up was also observed at the bottom of the flask in which the SCl2 was stored. It reacts exothermically
and violently with water.
When the distillation was nearly finished it was clear that we couldn’t properly extract the SOCl2 out of the SCl2. This is
most likely due to several reasons: First of all, since the boiling points of these two liquids is so similar, that separating them is difficult, and
we would need a larger amount of SOCl2 compared to SCl2 to fractionally distill them and collect pure SOCl2. The
mistake earlier was to use all of our SCl2, we should have just used a very small amount of SCl2 and a stoichiometric excess of
SO3.
So this is what I will do next: Use 1kg of Na2S2O8 to produce SO3 and distill it directly
into 50ml SCl2. In terms of stoichiometry, this should easily produce enough SO3 to turn all of the SCl2 into
SOCl2.
Question: Does anyone have an idea why this solidification in flask B happens when SO3 first drops into the
SCl2, and what this solid might be?
[Edited on 16-12-2021 by Monoamine]
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
| Quote: Originally posted by Lionel Spanner | ]
It mostly seems to be used for making chlorine-substituted thioethers from alkenes, S/N heterocycles from nitriles, and various polymeric species.
There needs to be a higher oxidation state at sulphur (IV or VI) to transfer chlorine without transferring the sulphur as well.
|
I see. Thank you for the insight Lionel Spanner!
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Successful 2 in 1 synthesis: Optimized home-synthesis of SOCl2. Phase I.
It worked! 
The previous attempt to make SOCl2 didn’t give a pure product, most likely because we had used far more SCl2 than
SO3, we will try this again but with a few improvements from the lessons we learned:
1) Distill the SO3 into the SCl2 directly. Don’t store it first and definitely don’t distill it into
H2SO4 first.
2) Use the 100% H2SO4 from the previous trials as catalyst. This is optional of course, but it should help to improve the
SO3 yield.
3) Grease all the joints with H2SO4 before starting the experiment!
4) Fill the receiving flask with much less SCl2 than the theoretical yield of SO4.
We will make SO4 from 1000g of Na2S2O8. If we use 100% H2SO4, then theoretically,
one mole of Na2S2O8 should produce one mole of SO3. So, since the molar mass of
Na2S2O8 is 238.03g/mol, we would expect to make 4.20 moles of SO3.
The density of SCl2 is about 1.62g/ml and its molar mass is 102.97g/mol. Therefore, 50ml of SCl2 should contain:
50ml * 1.62g/ml / 102.97g/mol = 1.57mol of SCl2
In other words, we should expect to have plenty of SO3 to convert all of the SCl2 into SOCl2.

A 1L round bottom flask (flask A in the previous schematic) was filled with 1kg of Na2S2O8 and 20ml of 100%
H2SO4. The receiving flask (flask B in the previous schematic) was filled with 50ml of SCl2. A Graham condenser was
fitted to flask B and to the end of the condenser was attached a hose leading into a pickle jar filled with NaOH (since my drying tube had become
clogged after the previous experiment). Neither condenser was cooled with water. Flask A was then wrapped in aluminum foil and the heating mantle was
cranked to the max and heated to around 300C.
As SO3 started coming over, the coloration of the SCl2 in the receiving flask became more and more translucent and approached a
much lighter orange.

This is to be expected as SO3 converts SCl2 into SOCl2, which is pale yellow to colourless. However, it was clear
that we weren’t generating enough SO3 to complete the reaction. This was almost certainly due to forming the SO3 at a
temperature that was too high (above 300C) and therefore distilling H2SO4 out of flask A, thus depriving the
Na2S2O7 of its catalyst.
Therefore, over the course of the experiment, 3x30ml of 100% H2SO4 were very slowly added to flask A, whenever we had cooled it
down enough that the temperature in flask A permitted the addition of more H2SO4. This did increase the drip rate, and also
helped to generate some additional SO3, but it was still not enough to fully convert all the SCl2 into SOCl2.
Instead, something very strange happened: Rather than becoming more and more translucent, the solution in flask B became darker and darker, going from
a dark orange to a blood red burgundy to almost black/brown/violet!
 
Nice coloration, but not what we want. Or is it...?
In the end, the process was stopped and we collected the dark liquid in the receiving flask.
At this point I thought that the experiment had failed and so I left everything there for two days. But when I went back in a few days later, the air
was thick with a delicious biting HCl smell – a very good sign!
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Successful 2 in 1 synthesis: Optimized home-synthesis of SOCl2. Phase II.
To my surprise, the colour in flask B from before had changed from a dark purple/brown to a very pale translucent orange!

A quick sniff with the nose confirmed that a strong reek of HCl (the telltale smell of SOCl2) was coming out of the flask . At this point I
strongly suspected that there was actually a decent amount of SOCl2 in the flask, and so I decided to give purification by fractional
distillation another try:

The flask with the pale orange liquid was set up for distillation (using the same setup as previously), but this time on an oil bath (both because
I’m having trouble with my electricity and because it allows for controlling the temperature more accurately).
As the internal temperature in the flask increased, a huge amount of HCl fumes evolved out of the apparatus and ate through the NaOH in the drying
tube in a few minutes, which was so exothermic that the small amount of moisture in the tube began to boil. I couldn’t breathe without a gas mask
anymore and just to be safe I held a bottle of 25% ammonia close by so I could visualize where the fumes were.
At about 80C internal temperature, the first drops of distillate came over at a pretty slow drip rate. The temperature was slowly increased to 90C and
then to 100C. Very quickly the drops changed from orange to (seemingly) clear, and so the bottles were swapped out since it was clear that no more
SCl2 was coming over.
Over the course of half an hour, the internal temperature was increased to about 150C, at which point a quick and steady drip rate had been achieved.

At this point it was also observed that no more HCl fumes evolved from the apparatus. Presumably the heavy HCl fuming earlier had been from
SOCl2 reacting with residual moisture in the apparatus, and so the lack of HCl fumes indicated that the apparatus was now completely dry.
We kept collecting distillate until no more boiling was observed in the reaction flask and no more distillate came over.
Pleasingly, only about 1-2ml of orange “stuff” (maybe S2Cl2?) remained in the reaction flask, and a similar amount of
SCl2 was likely in the first fraction that had been collected.
The rest was a translucent liquid with a slightly yellow tinge: SOCl2! In the end, about 75ml of SOCl2 were collected.
Considering that we started with 50ml of SCl2, this is a satisfactory result. More importantly it shows that an almost total conversion of
SCl2 had taken place!
1kg of Na2S2O8 with 110ml 100% H2SO4 (although this amount almost certainly can be reduced if
the temperature is controlled more patiently) together with 50ml of SCl2, can give 75ml of SOCl2.

|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Discussion
Well, it seems that this method works and is pretty efficient, in the sense that almost no SCl2 is wasted. The most resource-intensive part
is probably the 1kg of sodium persulfate, but fortunately this is also the cheapest ingredient (at least where I live). I also still believe that
someone with more skill than me can probably greatly increase the yield of SO3, further reducing the quantity of sodium persulfate that is
needed.
I now also think I can hazard a guess about my earlier question regarding the build up of solid in the flask B when SO3 is distilled into
SCl2. I think that this actually is mostly unreacted SO3. The reason for this guess is that, as was previously remarked, the
SOCl2 smell was only observed in flask B after two days and the colour had also changed dramatically from brown/purple to translucent
orange. I think that SO3 actually reacts with SCl2 very slowly, much much slower than I thought , and that it
first just dissolves into the SCl2 and then reacts with it over the course of a day or so. At least at ~0-10C ambient temperature (my shed
is pretty cold).
Finally, is this all worth it? Is this a viable way to obtain SOCl2 if you can’t source it commercially? Well, it is a multi-day
synthesis, but I think that now that I know how it’s done I can say that it can be done in about 2-3 days. Maybe 1 day for the SCl2
synthesis, half a day for the SO3/SCl2 mixture synthesis, and then (maybe the following weekend) the fractional distillation to
purify the SOCl2. Also, it should not be too difficult to scale this procedure up to produce a pretty sizeable amount of SOCl2
for only a small increase in cost, so in a few days of work I think you could definitely make a few hundred ml of SOCl2 like this.
If you just want a few deciliters of SOCl2 fast, then taking it out of batteries (like this) is probably the way to go, but if you want to make a few year’s worth supply, then using this procedure should be more efficient/cost
effective.
[Edited on 16-12-2021 by Monoamine]
[Edited on 16-12-2021 by Monoamine]
|
|
|
teodor
National Hazard
Posts: 876
Registered: 28-6-2019
Location: Heerenveen
Member Is Offline
|
|
| Quote: Originally posted by Monoamine | | I now also think I can hazard a guess about my earlier question regarding the build up of solid in the flask B when SO3 is distilled into
SCl2. I think that this actually is mostly unreacted SO3. |
Also, in the presence of air the sulfur oxytetrachloride, ClSO2 * SCl3 could be formed (mp = 57C) and this compound is disproportionating on standing:
ClSO3 * SCl3 -> SO2Cl2 + SOCl2
[Edited on 17-12-2021 by teodor]
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Hi teodor. Do you know how much air (do you mean O2?) is needed for this to occur? The reason I ask, is because the only time there is a
significant amount of O2 present is when the sodium persulfate decomposes into sodium pyrosulfate - which is long before any SO3
is distilled into the SCl2. Also, while there may be some trace SO2Cl2 in the final product, there would only be a
very small amount since SO2Cl2 decomposes at 100C and the distillation was carried out at 150C internal temperature.
(But maybe decomposing SO2Cl2 is what gave rise to the large amounts of HCl fumes that blew out of the apparatus at the
beginning of the fractional distillation?)
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Some afterthoughts
The thionyl chloride was sealed into glass ampules (made from test tubes) for long term storage. The reason for this is that SOCl2
has a tendency to leak out of most bottles it's in (like bromine). If storing in glass ampules is not an option, then a bottle with a glass
or PTFE cap put into a ziplock bag filled with dry Na2CO3 is also an option. The sodium carbonate acts as a dessicant and
neutralizes escaping acidic fumes, while releasing CO2, which blows up the ziplock bag and so gives a visual indication of when leakage has
occurred.

Some tests were performed on the SCl2. First, it was found that sulfuric acid and sulfur dichloride are not miscible.
(This really surprised me!)

Adding some phosphorus pentoxide (P2O5) to the test tube generated SO3 in-situ and so initiated a conversion of
SCl2 to SOCl2, which was greatly sped up when the test tube was heated a little.

The result is a bottom layer of H2SO4 and a top layer of SOCl2. The brown tinge is probably contaminants (maybe also
from the P2O5?)

Does anybody know why SOCl2 is immiscible with H2SO4?
|
|
|
Fery
International Hazard
Posts: 1015
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
I store SOCl2 in its original bottle, and yes it is permanently slowly leaking. I covered the stopper with few layers of aluminium foil which
neutralizes leaks of this nasty compound whicht eats through Al in few months so I have to check the bottle periodically and replace the Al foil.
Btw I suppose your H2SO4 in the bottom layer is 100% otherwise SOCl2 would react with all traces of H2O in your H2SO4. In fact you previously produced
oleum so there is no H2O. AFAIK there is no reaction between H2SO4 and SOCl2. SO3 could react with HCl into chlorosulfuric acid but you do not have
H2O there, so no HCl produced by hydrolysis of SOCl2.
[Edited on 19-12-2021 by Fery]
|
|
|
teodor
National Hazard
Posts: 876
Registered: 28-6-2019
Location: Heerenveen
Member Is Offline
|
|
| Quote: Originally posted by Monoamine | Hi teodor. Do you know how much air (do you mean O2?) is needed for this to occur? The reason I ask, is because the only time there is a
significant amount of O2 present is when the sodium persulfate decomposes into sodium pyrosulfate - which is long before any SO3
is distilled into the SCl2. Also, while there may be some trace SO2Cl2 in the final product, there would only be a
very small amount since SO2Cl2 decomposes at 100C and the distillation was carried out at 150C internal temperature.
(But maybe decomposing SO2Cl2 is what gave rise to the large amounts of HCl fumes that blew out of the apparatus at the
beginning of the fractional distillation?) |
Hi Monoamine.
No, I don't know. In one old book, it was mentioned that this compound is formed when chlorine, which is not free from O2, acts upon SCl2. But H2SO4
and SOCl2 or SO2Cl2 can also give HClSO3, which probably can react with SCl4 which probably could be formed by SCl2 disproportionation to S2Cl2 and
SCl4.
It's only a guess and I believe it could be far from the reality, especially if the solid compound is yellow because SClO3 * SCl3 should be colorless.
I know that the presence of as little as 0.2% of SOCl2 prevents the polymerization of SO3. It is used as a stabilizer of the liquid (alpha) form. On
the other hand, the presence of even traces of water (1 H2O molecule per 100000 SO3 molecules) cause polymerization. It would be interesting to see
what will be different if one adds SOCl2 to the pyrosulfate mixture. But water comes from H2SO4. Is it possible to use chlorosulfonic acid as a
catalyst for SO3 production here?
Your assumption that SO2Cl2 is decomposing could be wrong, because
2HClSO3 <-> SO2Cl2 + H2SO4
is an equilibrium at 170C. In this system, many complex reactions occur and the darkening of the mixture is some evidence of the presence of other
compounds, probably
with sulfur-sulfur bonds.
But the exact composition probably doesn't matter. The important step is disproportionation which gives sulfur in the 4th oxidation state starting
from 2 and 6, and I think this step is essential to get SOCl2, also it can go through some other intermediate compounds. You managed it exceptionally
well, so, congratulations!
[Edited on 21-12-2021 by teodor]
[Edited on 21-12-2021 by teodor]
[Edited on 21-12-2021 by teodor]
|
|
|
Monoamine
Hazard to Others
Posts: 168
Registered: 25-5-2021
Location: Sweden(ish)
Member Is Offline
Mood: +7
|
|
Very interesting idea to use chlorosulfonic acid as a catalyst teodor! I don't know if it also works, but it's certainly worth a try.
Now that I think about it, I'm sure that the final product contains some, since some of the HCl produced must have reacted with some of the
SO3 or even H2SO4. Also, if a drop of the final product was placed on paper it immediately blackened it, like oleum.
I wonder if sulfur dichloride would also react with chlorosulfonic acid to make thionyl chloride? But I assume that it would, since chlorosulfonic
acid is almost certainly an intermediate of sorts (SOCl2 and SO3 would produce it). As you say, there are probably many
additional reactions going on at the same time, and SOCl2 is likely the energetically most favourable product, which is why the reaction
tends towards it eventually.
|
|
|
teodor
National Hazard
Posts: 876
Registered: 28-6-2019
Location: Heerenveen
Member Is Offline
|
|
Monoamine,
I have enough quantity of SOCl2 and that is the only reason I don't try to repeat your experiment which is very interesting indeed.
I think you can try to remove chlorosulfonic acid with NaCl or follow a procedure of SOCl2 purification with quinoline or dimethylaniline which I also
want to try because for some applications it is better to use a purified compound.
I have an impression, just from the theoretical study of the topic, that SOCl2 in SO3/SCl2 system is not favorable at all and this is why there are
only 2 reports of successful synthesis on SM. And it looks like the synthesis has some peculiar point which I think is a disproportionation reaction
when some intermediate compound results in SOCl2/SO2Cl2 mixture.
It is different when SO2 is used because it looks like in this case SOCl2 is formed in a pure form. I mean the reaction of SO2 (usually in liquid
form) with PCl5, NbCl5, WCl6, and similar higher chlorides.
|
|
|
Fery
International Hazard
Posts: 1015
Registered: 27-8-2019
Location: Czechoslovakia
Member Is Offline
|
|
Hi teodor, I have plenty of quinoline so I can offer it to you for free. More than 10 small bottles of 100 ml then IIRC 1x 1 L bottle and also maybe 1
or 2 bottles of volumes like 250 ml and 500 ml. It is just a little yellow-brown, not colorless anymore. It would require vacuum distillation. All
these bottles are somewhat old like 10-30 years. SOCl2 is so nasty that I'm quite afraid to purify it. Whenever possible I would prefer to use it
directly from original bottle. Which syntheses require very pure SOCl2?
|
|
|
teodor
National Hazard
Posts: 876
Registered: 28-6-2019
Location: Heerenveen
Member Is Offline
|
|
Yes, Fery, SOCl2 is nasty, for that reason, I will try the distillation after improving my fume hood. There are 2 things I plan to do: change the fan
and make additional air inlets from the bottom and behind. I can detect SOCl2 leaks easily but for some other toxic compound the leak detection could
be harder, so I prefer to practice with nasty SOCl2 first. If I will manage it I will be able to distill other similarly toxic things in my hood.
If you can give me some quinoline for the distillation it would be great, thank you very much.
I prefer to use pure compounds in many types of reactions, especially when I do experiments or synthesis which I didn't make before, to put the
reference in the bottle, then I will be able to compare any other results with my references.
|
|
|
| Pages:
1
2 |












