Austin543
Harmless

Posts: 2
Registered: 28-7-2019
Location: US
Member Is Offline
Mood: Saturated
|
|
Pyrazine Synthesis?
A few months ago, I stumbled upon a paper that described some copper complexes that exhibit a wide range of fluorescent colors, almost all the colors
in the rainbow in fact (https://pubs.acs.org/doi/abs/10.1021/ic0510359). However, one of the major ligands used is pyrazine or its derivatives. After doing some research
I've realized that such a simple molecule is a lot harder to synthesize than I would've imagined.
I've come across quite a few syntheses, all of which have disadvantages. The first one starts with ethyl acetoacetate and it seems like the best
method so far, however with many steps comes a reduced yield. I've posted the the pathway and the paper that it's from below. It forms
dimethylpyrazine, but according to another source I found, KMnO4 can be used to oxidize the methyl groups yielding a carboxylic pyrazine (bottom of page 8). In that source they
oxidized tetramethylpyrazine, but I don't see why it wouldn't do the same for the dimethyl version. From there you could do a decarboxylation similar
to the common pyridine synthesis from niacin (pyridine-3-carboxylic acid).
Another route involves the gas phase reaction of ethylene diamine over a copper catalyst. They mention copper chromite which is easily prepared as
shown by Doug's Lab, however it is a gas phase reaction, which makes the apparatus quite a bit more complicated. My idea for the apparatus would be
passing ethylene diamine vapor over a bed of catalyst similar to how Nile Red made SiCl4, but who knows how that would turn out.
I also thought of doing a simple ring forming reaction similar to the dioxane synthesis from ethylene glycol. Using glyoxal and ethylene diamine, but
it seems that it might make piperazine-2,3-diol among other things and sources are questionable.
Anyway, that's everything that I've come across regarding a piperazine synthesis and I wanted to hear your thoughts and see if any of you guys have
tried to make it before or know a better method 
Attachment: pyrazine from ethyl acetoacetate.pdf (212kB)
This file has been downloaded 480 times
|
|
|
Pumukli
National Hazard
Posts: 705
Registered: 2-3-2014
Location: EU
Member Is Online
Mood: No Mood
|
|
I doubt if those two methyl groups would make a big difference as a ligand. Just do the synthesis and report what you find! :-)
Even if failed to luminesce the dimethyl-pyrazine synthesis in itself would be a worthy reading. I think no one attempted such a synth on
science-madness so far.
I can see that it was your first post, so welcome in the group. We need more budding amateur organic chemists, especially the ambitious type. ;-)
|
|
|
Σldritch
Hazard to Others
Posts: 309
Registered: 22-3-2016
Member Is Offline
Mood: No Mood
|
|
2,5-dimethylpyrazine route
I had a great idea how to make 2,5-dimethylpyrazine. It may even be possible as a one pot mix and leave synthesis but im not sure if the acetone
oxidation works in ammonia nor under what conditions the oxidation to pyrazine works. There should be a thread with more information on the acetone
oxidation somewhere on the forum. As for the oxidation to pyrazine, copper is a pretty good air oxidation catalyst in aqeous solution, even oxidizing
ammonia to nitrate. It may even be possible to cheap out on Copper Chloride by using it as a catalyst in step one with air. Workup should be a simple
distillation i believe, unless the presence of byproducts does not make it foam horribly (it probably will).
[Edited on 29-7-2020 by Σldritch]

|
|
|
Austin543
Harmless
Posts: 2
Registered: 28-7-2019
Location: US
Member Is Offline
Mood: Saturated
|
|
| Quote: |
I doubt if those two methyl groups would make a big difference as a ligand. Just do the synthesis and report what you find! :-)
|
Thats a good point! It seems like most of these syntheses give dimethyl or trimethyl pyrazine so hopefully skipping the oxidation step would work.
| Quote: |
I had a great idea how to make 2,5-dimethylpyrazine. It may even be possible as a one pot mix and leave synthesis but im not sure if the acetone
oxidation works in ammonia nor under what conditions the oxidation to pyrazine works. There should be a thread with more information on the acetone
oxidation somewhere on the forum. As for the oxidation to pyrazine, copper is a pretty good air oxidation catalyst in aqeous solution, even oxidizing
ammonia to nitrate. It may even be possible to cheap out on Copper Chloride by using it as a catalyst in step one with air. Workup should be a simple
distillation i believe, unless the presence of byproducts does not make it foam horribly (it probably will). |
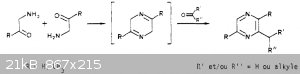
I did some research and found a french paper that describes what you're talking about with the aminoacetone but it says you'd also need to react it with a carbonyl compound in
order to isolate the cyclized ring. If you used formaldehyde it seems like you'd get trimethylpyrazine, which hopefully wont affect florescence, but
if it does I can try the permanganate oxidation step.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
2,3-Dimethylpyrazine synthesis
From my literature searching I figured the direct synthesis of dihydropyrazine from glyoxal and ethylene diamine would not work and produce other
compounds, see J. M. Edwards et al., Chem. Commun. (London) 1968, 1649-1650, https://doi.org/10.1039/C19680001649:
Attachment: edwards1968_glyoxal_ethylenediamine_cage_formation.pdf (117kB)
This file has been downloaded 240 times
However, diacetyl should work and indeed it does, producing a 2,3-dimethyl-5,6-dihydropyrazine which on oxidation can form 2,3-dimethylpyrazine.
Oxidation with permanganate apparently produces 3-methylpyrazine-2-carboxylic acid fairly easily, which can decarboxylate to give 2-methylpyrazine.
But I am sure more harsh oxidation conditions could provide the dicarboxylic acid, which again easily decarboxylates.
Diacetyl was obtained as a perfume ingredient, but it seems to be no longer sold where I bought it. It has a lovely buttery aroma and is a yellow
liquid. It should also be possible to make by using methylethylketone and a nitrite ester, then hydrolysing the diacetylmonooxime obtained.
Ethylene diamine was obtained from a chemical supplier.
The first step of the procedure was taken from T. Ishiguro, M. Matsumura, Yakugaku Zasshi 1958, 78, 3, 229-231, https://doi.org/10.1248/yakushi1947.78.3_229:
Attachment: matsumura1947_dimethyldihydropyrazine.pdf (545kB)
This file has been downloaded 286 times
However, I deviated from their synthesis, which may have contributed to the low yield. I will repeat this at a later date.
Also of value was F. Jorre, "Synthese von 2,3-Dimethylpyrazin", Dissertation, Kiel 1897, urn:nbn:de:gbv:8:2-2889314. It's freely available at the link provided, but is too big to be attached here.
The oxidation was performed according to T. Akiyama, J. Agric. Food Chem. 1978, 26, 5, 1176-1179, https://doi.org/10.1021/jf60219a057:
Attachment: akiyama1978_dihydropyrazine_oxidation.pdf (415kB)
This file has been downloaded 247 times
Again, I deviated and should have used proper manganese dioxide and not battery crap, and also I should not have used denatured alcohol as there were
aldol side reactions.
2,3-Dimethylpyrazine has an entry in Beilsteins Handbuch der organischen Chemie, 4th edition, book 23 (1936), p. 95, available on archive.org here.
Experimental:
1.42 g of ethylene diamine (the paper uses 1.85 g of the hydrate, I used anhydrous) and 20 mL of diethyl ether were stirred in a 100 mL RBF in an
icebath. After it had cooled, a solution of 2.00 g of diacetly and 10 mL of Et2O was added dropwise via an addition funnel during 20 minutes. There is
a white precipitate appearing almost immediately. The diacetyl solution on the glass walls above the liquid turns a red color, but the solution itself
is almost colorless. After complete addition the ice bath was removed and the addition funnel replaced with a reflux condenser. It was stirred at room
temp. for 1 hour. The procedure stated to stir until it goes clear, which I didn't do. Then the solution was refluxed on a water bath for 30 minutes,
during which it cleared up. A slightly yellow ether layer and a reddish bottom layer was visible. 2.00 g of powdered KOH was added and the solution
stirred for 1 hour. The ether becomes more yellow.
Here I deviated from the procedure. The procedure now removes the ether layer and distills it under vacuum to obtain the
2,3-dimethyl-5,6-dihydropyrazine. Since I needed KOH for the next step anyways I figured I could get away without this. It might be part of the reason
for the bad yield.
The mixture of ether and KOH was distilled in a hot water bath of 50-60 °C until most of the ether, around 20 mL, was removed.
Then 40 mL of 95% EtOH (denatured but this is a terrible idea) and 12 g of crude manganese dioxide (again, use a higher grade) was added and the
solution refluxed for 4 hours. After cooling, filtration by gravity was extremely slow, but filtration by vacuum acceptable. The residue was washed
with 4x 10 mL of EtOH. The solution is dark red (presumably aldol side products). The EtOH was distilled off until only ~5 mL remained in the flask,
to which 10 mL of sat. NaCl-solution was added. This is extracted 2x with 10 mL of Et2O (I recommend doing more extractions). The yellow Et2O is
retained.
The Et2O extract is distilled and the ether collected. Distillate was obtained at 156-160 °C (large range due to small scale and large apparatus) and
amounts to 0.2755 g (11%).

The aqueous solution has a pH of 7-8, shows blue fluorescence and has a stale odor.
The boiling point of the 2,3-dimethyl-5,6-dihydropyrazine is reported as 162-165 °C, the 2,3-dimethylpyrazine as 156 °C. The solubility of the
2,3-dimethyl-5,6-dihydropyrazine is high in water and ethanol but much lower in ether, the solubility of the 2,3-dimethylpyrazine is high in water,
alcohol and ether. The 2,3-dimethyl-5,6-dihydropyrazine is reported to show a strongly basic reaction in water, the 2,3-dimethylpyrazine is supposed
to react neutral.
It is also of interest that the 2,3-dimethylpyrazine is steam volatile.
Overall this leaves much to be improved, but I will give this another attempt.
[Edited on 6-9-2022 by Diachrynic]
[Edited on 6-9-2022 by Diachrynic]
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Interesting synthesis Diachrynic, I am interested in pyrazine compounds too. I would be interested to hear how you get on if you have another go. One
point is that using MnO2 as an oxidizing agent in a strongly alkaline situation seems strange. Usually there is at least some acetic acid added to
remove the Mn2+ formed and keep the reaction chugging forward. In an alkaline medium where does the Mn2+ go? Or do you end up with an Mn-pyrazine
complex (the red colour?).
Some years ago I found saw 500g of piperazine hydrate for sale on ebay and bought it. My cunning plan is to try and oxidise it to pyrazine or perhaps
to try to de-hydrogenate it with palladium or raney nickel. I don't know if this would work and I haven't had time to try it yet.
By the way that German dissertation; can it be downloaded in one go or is it just page by page?
|
|
|
Lionel Spanner
Hazard to Others
Posts: 168
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
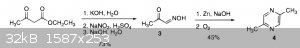
The first scheme in the OP is a Gutknecht synthesis, which is a good general method for making pyrazines that are inversely symmetrical about the
nitrogens. If memory serves, the second step in the original reaction reduced the oxime using tin and hydrochloric acid as opposed to zinc and caustic
soda - not sure if this would improve the yield.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Quote: Originally posted by Boffis  | | One point is that using MnO2 as an oxidizing agent in a strongly alkaline situation seems strange. Usually there is at least some acetic acid added to
remove the Mn2+ formed and keep the reaction chugging forward. In an alkaline medium where does the Mn2+ go? Or do you end up with an Mn-pyrazine
complex (the red colour?). |
I assume that Mn2O3 is formed, but the paper doesn't talk about it. The reaction mixture after filtration was indeed red, but I
assume that is merely from aldol condensations from using denatured ethanol since Mn(II) is not stable in basic conditions I think. A rerun using
ketone-free alcohol should tell if this assertion is correct or not.
Good luck on the piperazine oxidation, I got the impression that piperazine is fairly hard to aromatize while searching for pyrazine procedures. But
then again I never looked too much into it since I don't have any. Piperazine citrate, a dewormer that used to be OTC in some places, seems to be
phased out by now.
It can be downloaded in sections, but I haven't found the button to download it all at once. Navigate into the section you want, then go to "Zitieren
und Nachnutzen" > "Kapitel" > "PDF". It should redirect you to the following links: Title page, introduction, experimental, curriculum vitae.
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Many thanks Diachrynic; I successfully downloaded the thesis.
By the way have you seen "the chemistry of heterocyclic compounds" series, specifically volume 41 Pyrazines? They can be downloaded from Libgen but
its a big file, about 28 MB. Apparently it is possible to dehydrogenate piperazine to pyrazine by distillation with lime and zinc dust; sounds brutal
and inefficient but I might try something like MnO2 and acetic acid or lime and sulphur.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Thank you for the book recommendation!
| Quote: Originally posted by Boffis | | Apparently it is possible to dehydrogenate piperazine to pyrazine by distillation with lime and zinc dust; sounds brutal and inefficient but I might
try something like MnO2 and acetic acid or lime and sulphur. |
I was reading in Houben-Weyl
IV/1a p.125 today and found a reference for the preparation of pyrazine from piperazine using CuO at 300 °C with a yield of 94%.
The reference is S. P. Yurel, A. A. Anderson, M. V. Shimanskaya, Khimiya Geterotsiklicheskikh Soedinenii 1974, 1414-1419.
This is available in translation: S. P. Yurel, A. A. Anderson, M. V. Shimanskaya, Chemistry of Heterocyclic Compounds 1974,
10, 1241–1245, https://doi.org/10.1007/BF00470173.
Attachment: BF00470173_pyrazine_from_piperazine.pdf (403kB)
This file has been downloaded 218 times
I thought you might be interested.
While reading another large review article (Y. Yamamoto et al., Science Of Synthesis, Hetarenes and Related Ring Systems, Product Class 14,
Pyrazines 2014, 751, https://doi.org/10.1055/sos-SD-016-00919) I came across the mention of the reaction of 2,3-diaminopropionic acid with glyoxal, followed by air
oxidation, to give pyrazine-2-carboxylic acid. A procedure for this can be found in british patent GB1016468A:
Attachment: GB1016468A_pyrazine_carboxylic_acid_diaminopropionic_acid.pdf (333kB)
This file has been downloaded 221 times
Now 2,3-diaminopropionic acid isn't easy to make in itself. I had plans for this some time ago as a potential way to make ethylene diamine. Briefly,
alanine would be chlorinated in concentrated sulfuric acid to produce 3-chloro-2-aminopropionic acid (J. Kollonitsch, A. Rosegay, G. Doldouras, J.
Am. Chem. Soc. 1964, 86, 9, 1857-1858, https://doi.org/10.1021/ja01063a045) which should then react in some way with an excess of concentrated ammonia (which is presumably the nail in
the coffin for this reaction - the literature uses very concentrated ammonia and uses glass tubes, melted shut, to withstand the pressure when the
mixture is heated...) to form 2,3-diaminopropionic acid. The low solubility of its oxalate could possibly be exploited for purification. (E. Klebs,
Z. Physiol. Chem. 1894, 19, 22, https://doi.org/10.1515/bchm1.1895.19.4-5.301). But unless a less suicidal way of doing the substitution is found this won't be viable.
Another approach would be to use fumaric or maleic acid, dibrominate it and replace the bromides with amines (either maybe via ammonia or in two steps
using benzyl amine, followed by deprotection with hydrogen and Pd/C, for this way see W. Wenner, J. Org. Chem. 1948, 13, 1,
26-30, https://doi.org/10.1021/jo01159a004 and H. McKennis, A. S. Yard, J. Org. Chem. 1958, 23, 7, 980-982, https://doi.org/10.1021/jo01101a010). This provides 2,3-diaminosuccinic acid which can condense with glyoxal in much the same way as mentioned
above to form pyrazine-2,3-dicarboxylic acid. (A. V. R. Rao et al, Indian J. Chem., Sect. B 1984, 9, 850, I could only find
the ChemInform version under https://doi.org/10.1002/chin.198452235.)
I looked more into the literature and here are the methods for oxidation of dihydropyrazines to pyrazines which I have found so far:
FeCl3 in EtOH (WO2000025791A1; P. J. Steel, G. B. Caygill, J. Organometallic Chem. 1990, 395, 359-373, https://doi.org/10.1016/0022-328X(90)85299-E)
DDQ (A-C. Chen, Tetrahedron 2008, 64, 37, 8907-8921, https://doi.org/10.1016/j.tet.2008.06.054)
Nickel peroxide (T. Kobayashi, J. Chem. Soc., Perkin Trans. 1 2001, 12, 1372-1385, https://doi.org/10.1039/B101330K)
NaOEt in MeOH under N2 (What is going on with this one?) (Experimental of compound 16 in M. Isbera et al., Synthesis
2019, 51, 23, 4463-4472, https://doi.org/10.1055/s-0039-1690678)
KOH, EtOH, O2 (C. M. Fitchett, P. J. Steel, ARKIVOC (Gainesville, FL, U. S.) 2005, 218-225, https://doi.org/10.3998/ark.5550190.0007.318)
Sulfur (A. Ohta, J. Heterocyclic Chem. 1984, 21, 103, https://doi.org/10.1002/jhet.5570210122)
MnO2, CuO (T. Akiyama, J. Agric. Food Chem. 1978, 26, 5, 1176-1179, https://doi.org/10.1021/jf60219a057)
Fehlings reagent, potassium ferricyanide (F. Jorre, Dissertation, Kiel 1897, urn:nbn:de:gbv:8:2-2889314)
Chloranil (This supposedly gives near quantitative yield, is much faster than MnO2, and uses stoichometric amounts: N. Furuta,
Heterocycles 2009, 77, 2, 1079-1088, https://doi.org/10.3987/com-08-s(f)87) (Axt has prepared chloranil from paracetamol, having done this myself I should warn that the reaction is indeed vigourous, foams a lot, and makes a cloud of
nitrogen dioxide, if the nitric acid is added at once.)
HNO3 in AcOH (The derivative is described as being very easy to oxidize so this may not generalize, A. Hildesheimer, Ber. Dtsch.
Chem. Ges. 1910, 43, 3, 2796-2805, https://doi.org/10.1002/cber.19100430338)
HNO2 (Again, this substrate seems easy to oxidize, S. Gabriel, Ber. Dtsch. Chem. Ges. 1908, 41, 1, 1134, https://doi.org/10.1002/cber.190804101223)
Some seem to be easier to oxidize than others (it seems especially phenyl substituted ones) so not all of these methods will work for the ones at
hand. But maybe there are some useable ones that are less messy than the ones already mentioned.
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Diachrynic, have you moved along on this project?
I have been doing some experimentation myself I am currently steam distilling some 2,3-dimethylpyrazine, made by ferricyanide of ethylenediamine and
biacetyl in water. I carried out the oxidation almost immediately. The aqueous reaction mixture turns brown very quickly and heats up. I used only
cold tap water for cooling. If I do this again I may try an ice bath. The ferricyanide oxidation I rapid and give less solution than the fehling's
solution route.
I have also prepared some 2,3-dimethylquinoxaline which I am going to try oxidizing with permanganate. I have also prepared a batch of glyoxal
bisulphite complex for the preparation of un-substituted quinoxaline and hence to pyrazine dicarboxylic acid via permanganate oxidation again.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Hey Boffis, no, sadly I had very little time on my hands recently to revisit this. Other things took priority. I have prepared some MnO2 ·
2 H2O per NurdRage's procedure however.
But I did notice that the distillate in the ampoule has taken on a slight yellowish color. Presumably it isn't quite as pure - not too really much of
a surprise because of the small amount in too large of an apparatus. I am also waiting for some glassware to arrive to be able to do smaller scale
distillation.
How did the oxidation with ferricyanide go? Were you able to isolate a decent yield? It would be quite convenient if this gave decent yields.
| Quote: Originally posted by Boffis | | I have also prepared a batch of glyoxal bisulphite complex for the preparation of un-substituted quinoxaline and hence to pyrazine dicarboxylic acid
via permanganate oxidation again. |
Very cool! What I was unhappy about is that a large excess of permangante is required. (Example: 5.8 eq. in C. A. Obafemi, W. Pfleiderer, Helv.
Chim. Acta 1994, 77(6), 1549–1556, https://doi.org/10.1002/hlca.19940770610)
Also, phenylene diamine isn't easy to find for me.
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Diachrynic, The dark brown reaction product from the reaction of biacetyl with ethylenediamine looked very unpromising but I mixed it with the 2
Molar equivalence of potassium ferricyanide required and got a dark brown liquid that smelled of musty roast chicken . I then added some extra water and distilled it until the distillate no longer
smelled of roast chicken. So far so good.
The problem then arises that the original paper suggests isolation by precipitation of the mercuric chloride complex formed by adding saturated
mercuric chloride solution (about 6% at RT) but since each g of product requires about 5g of mercuric chloride (say 85ml of solution) and my
theoretical yield was about 11g.... that's shit loads of mercuric chloride solution! A small scale experiment certainly gave a nearly white ppt on
addition of mercuric chloride but it slowly turned orange (=mercuric oxide?) and then grey on drying the ppt. I think I will try ether extraction and
then proceed as you did, tomorrow. Interestingly after the oxidation masses of potassium ferrocyanide crystallised out and after distillation I
recovered more. Out of a theoretical amount of 83g of K ferrocyanide 3 hydrate I recovered 75g. I recrystallised this with charcoal treatment to
obtain 62g of clean yellow K ferrocyanide crystals which I am now going to ty and oxide back to ferricyanide with chlorine. So the evidence so far is
that ferricyanide in alkaline conditions works. The losses are probably in the initial condensation which looks messy.
Now I have prepared the glyoxal sodium bisulphite compound I am going to try this with biacetyl and then condense it with ethylenediamine. My hope is
that with ethylenediamine the reaction will be slower and less messy, less dark brown aldol type crap. This technique certainly works with the
o-phenylenediamine condensation with glyoxal when the yield rises from 30 to 90%.
Actually the oxidation of quinoxaline to pyrazine-2,3-dicarboxylic acid theoretically requires 6 eq. of KMnO4!!
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
A quick up-date; I tried the ether extraction of the remaining oxidized liquor that hopefully contained the 2,3-dimethylpyrazine. I extracted the
liquor (about 200ml) 5 x with 30ml portions of ether. The extracts were combined, dried with anhydrous magnesium sulphate and most of the ether
distilled off using a warm hotplate. The final liquor, about 15ml which had already begun to deposit crystals was poured into a glass bowl and allowed
to evaporate. A small crop of nicely colourless crystals has formed, currently drying. I will weigh them tomorrow but looks like <0.2g so about 2%
yield! Mmmmm could do better! but I need to investigate the product.
Final yield turned out to be 0.601g so 6% yield. Still shit!
[Edited on 25-10-2022 by Boffis]
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
| Quote: Originally posted by Boffis | | The final liquor, about 15ml which had already begun to deposit crystals was poured into a glass bowl and allowed to evaporate. A small crop of
nicely colourless crystals has formed, currently drying. |
This is concerning as 2,3-dimethylpyrazine is a liquid at room temperature! Beilstein and Jorre both state that it stays liquid at 0 °C, while a more
recent paper (R. Boese et al., Acta Crystallogr., Sect. B: Struct. Sci. 2000, 56(4), 677–681, https://doi.org/10.1107/s0108768199016547) states the melting point to be ~10 °C.
Please determine the melting point of your product! For example, the N-oxide melts at 85-86 °C, the N,N'-dioxide at 209-210 °C (C.
Sakuma et al., Magn. Reson. Chem. 1996, 34(7), 567–570, https://doi.org/10.1002/(SICI)1097-458X(199607)34:7<567::...).
But maybe something completely different is going on... Strange. And a shame about the yield, too.
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Hi Diachrynic; the Mp issue was rather my point about investigating my material. Curiously the crystals has a nice savoury small though in high
concentration it slightly irritates the nose so it has the right smell about it. The first yellowish crystals that crystallised from the ether are the
left over mercury 2,3-dimethylpyrazine and are odourless. No liquid was left on evaporation of the ether. The crystals have a Mp >45 because I
dried them at that temperature, they are also volatile which may in part account for the low yield.
I decided to reinvestigate the first stage of the reaction where the formation of much brown material seems to be at the expense of the product. I
recently prepared some sodium glyoxal bisulphite compound for the preparation of quinoxaline (benzopyrazine). It proved very easy to prepare and in
very high yield so I tried substituting biacetyl for glyoxal but no crystalline product formed in spite of the fact that the smell of SO2 practically
disappeared. So I am presuming that the compound is much more soluble than the glyoxal analog. Anyway I finally got fed up and simply added the
ethylenediamine to the biacetyl sulphite compound solution. The beaker became warm and a strong earthy roast meat aroma appeared but no brown tar.
Sounds promising so far but how do you separate the dihydro compound from the bisulphite solution? I may try adding salt and then extraction with
ether. With so much bisulphite present direct oxidation is not going to be possible.
I also tried the permanaganate oxidation of 2,3-dimethyl quinoxaline and it basically appears to have worked but the resulting
5,6-Dimethylpyrazine-2,3-dicarboxylic acid appears to very rather susceptible to oxidation and turns brown as fast as I can purify it. But this is
work in progress and I did the trial run on the discoloured material recovered from the residual solvent after distilling to recover the alcohol
solvent.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Very strange.
I am currently retrying the reaction, this time with isolation of the 2,3-dimethyl-5,6-dihydropyrazine. The crude yield of that intermediate is <
30% currently. A lot of tar formation in the reaction flask and left after the vacuum distillation.
I'm not quite sure why. Maybe I should add a stoichiometric amount of water to the ethylene diamine so that it matches the use of the hydrate in the
source paper? That would be a possible modification - don't really see how that would help, but maybe it would make less tar.
[EDIT]
So I did my previous experiment on twice the scale, got a yield of about 1.51 g (~29%) on the 2,3-dimethyl-5,6-dihydropyrazine after a vacuum
distillation (about 200 mbar, bp. ~110 °C). The oxidation was done in absolute undenatured ethanol with manganese dioxide dihydrate (prepared as per
NurdRage) was accompanied by a visible color change to brown in the oxide this time. Same workup as before, this time I extracted 4 times with ether
however. Finally, vacuum distillation yielded 0.365 g with a huge boiling range somewhere above 90 °C (~200 mbar) (It should be closer to 105 °C
again though!).
That is a yield of 7% at the very most - exceedingly disappointing.
[Edited on 31-10-2022 by Diachrynic]
we apologize for the inconvenience
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
I repeated the experiment in the last post, i.e. twice my original scale again, this time with enough added water to form ethylene diamine
monohydrate. It's not actually soluble in ether - as opposed to the anhydrous version, as Jorre noted correctly. I only added 1 g of KOH this time and
extracted the residue multiple times with ether. That residue I also extracted multiple times with DCM which seems to take up the coloration a lot
more readily.
The DCM idea was roughly based on the usage of chloroform in: I. Flament, P. Sonnay, G. Ohloff, Bull. Soc. Chim. Belges
1979, 88, 941, https://doi.org/10.1002/bscb.19790881115. (Thank you Ormarion for the translation.) Note that they use anhydrous ethylene diamine and also observe
this two phase system! They extract the lower phase with chloroform, and then distill the combined organic phases.
On distilling off the ether I was left with 6.60 g of orange liquid. The DCM only produced a small amount of solid gunk. Interestingly enough, another
drop of yellow smelly oil was obtained when letting the distilled DCM evaporate - maybe it "steam distilled" or codistilled with the DCM?
The 6.60 g were used without distillation and treated with KOH and manganese dioxide (dihydrate, again) in absolute ethanol. I distilled nearly all
the ethanol from the filtrate after the reaction, and extracted six times with ether. On distilling in the end I obtained a fraction at 155-158 °C,
amounting to 0.43 g of yellow oil. It should be colorless, and even if it were pure, it's only 8% of the theory.
I am at a loss how the literature reports 70-80% for each of the two steps. I have the feeling that the oxidation especially isn't working as it
should. I am getting tarry side products at every step.
we apologize for the inconvenience
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
2,3,5,6-Tetramethylpyrazine
Due to frustration with the synthesis of 2,3-dimethylpyrazine I decided to try the Gutknecht synthesis of 2,3,5,6-tetramethylpyrazine.
Comparing to literature yields it went rather poorly. It should also be noted that this compound is probably unable to form complexes[1]
like unsubstituted pyrazine does in the paper from the opening post.
There are several things to improve here. I decided to save myself a steam distillation and crystallization because of a shortage of time, but maybe
it would improve the yield. The end product also isn’t pure yet, so a recrystallization would probably be required. Maybe a different approach to
the reduction would improve the yield, like using tin and hydrochloric acid[6] or with tin(II) chloride and hydrochloric
acid.[7,8] The literature claims a quantitative yield for this approach,[2] but that clearly didn’t work out like that for me.
Also because of the close melting points of diacetylmonoxime and hydrated tetramethylpyrazine I can't actually be sure that it
worked.
Chemicals:
Methyl ethyl ketone (obtained from a chemical supplier)
Isopropyl nitrite (synthesized per Dougs Lab[9])
HCl (37%, obtained from a chemical supplier)
Zinc powder (<40 µm, 99.995%)
Sodium hydroxide (food grade)
Diethyl ether (distilled from starter fluid)
Diacetylmonoxime:[3,4]
The synthesis starts with diacetylmonoxime from methyl ethyl ketone (MEK) and isopropyl nitrite (iPrONO) using concentrated HCl as a catalyst. Into a
three necked, 100 mL RBF hanging in a 40 °C water bath with a thermometer, a pressure equalized addition funnel and a reflux condenser was added a
stir bar, 0.5 mL of concentrated HCl and 8.09 g of MEK. To the addition funnel were added 10 g of iPrONO. The drop rate was adjusted such that the
internal temperature was maintained in the range of 40-55 °C. An ice cube had to be used to cool the water bath during the addition. The addition
took 20 min, then the reaction was stirred for 40 min at 55 °C.

The reaction mixture.
Then the water bath was removed and the reaction mixture heated on the hotplate until the solution reached 90 °C where it was kept for 5 min. The
required temperature was reached with almost no delay. The flask was taken off heat and allowed to cool to 20 °C.
2,3,5,6-Tetramethylpyrazine:[2]
A solution of 2.50 g of NaOH in 20 mL water was prepared in an 800 mL beaker and 21 g of zinc powder were weighed out and put to the side. To the
sodium hydroxide solution the reaction mixture was poured, resulting in a color change to red. This immediate usage was based on a remark in [5] that
claimed it's easier to directly use these raw solutions. I was lazy and wanted to save myself another steam distillation. Then the zinc was added at
once as well as 10 more mL of water. The reaction mixture was stirred with only slight exotherm, then it was boiled for 20 min. The color changed to a
greenish-brown.
Afterwards, the solution was decanted while hot and the zinc powder washed with 10 mL and then 5 mL of boiling water. The water was extracted after
cooling to room temperature with a total of 40 mL of diethyl ether in 6 portions.
The ether was then removed by simple distillation. The red liquid freezes into an orange solid.

The orange solid.
About 30 mL of water were added and the mixture was steam distilled.
From the initial steam distillate crystals began to form. More steam distillate was collected separately, but nothing crystallized from it. The formed
crystals were filtered off and the filtrate combined with the other steam distillate, which was then extracted with a total of 40 mL of ether in 6
portions. The ether was distilled off, afterwards a slight vacuum (about 200-250 mbar) was applied shortly to remove the last traces of ether and a
slightly yellow liquid was obtained, which rapidly crystallized on standing.
 
The initial precipitated solid.

The extracted sample.
The yield of the sample from water was 0.11 g, the yield after the ether distillation was 1.17 g. In total 1.28 g was obtained, which would be a yield
of 17%.
The melting point of the two fractions was measured. The first melted below 55 °C, it was probably still very wet. The second fraction melted at
75-76.5 °C. The literature melting point for the hydrate is 75-78 °C, for the anhydrous form (formed after extended drying over calcium chloride) is
86-87 °C.[2]
Unfortunately the melting point of diacetylmonoxime is 76-76.5 °C[3,4] so the two are very difficult to distinguish by melting point.
I would only be able to say what I got after some more tests, but I don't have the time at the moment. The melting point is suspiciously fit for
diacetylmonoxime. Maybe it would change after extensive drying. Maybe there is some TLC system that can resolve it? Maybe diacetylmonoxime forms some
sort of transition metal complex that tells it apart from 2,3,5,6-tetramethylpyrazine? Or maybe it will just have decomposed until I have time in the
lab again as diacetylmonoxime is apparently unstable in storage.[3,4]
[1] - C. Navas, M. M. Turnbull, C. Giogas, C. P. Landee, W. Zhang, G. Pon, R. Willett, Polyhedron 1993, 12, 1019, https://doi.org/10.1016/S0277-5387(00)87178-8
[2] - O. Wallach, Nachrichten von der Gesellschaft der Wissenschaften zu Göttingen, Mathematisch-Physikalische Klasse 1927,
238, https://resolver.sub.uni-goettingen.de/purl?PPN252457811_192...
[3] - W. L. Semon, V. R. Damerell, J. Am. Chem. Soc. 1925, 47, 2033, https://doi.org/10.1021/ja01684a037
[4] - W. L. Semon, V. R. Damerell, Org. Synth. 1930, 10, 22, https://doi.org/10.15227/orgsyn.010.0022
[5] - I. J. Krems, P. E. Spoerri, Chemical reviews 1947, 40, 279, https://doi.org/10.1021/cr60126a004
[6] - H. Gutknecht, Ber. Dtsch. Chem. Ges. 1879, 12, 2290, https://doi.org/10.1002/cber.187901202284
[7] - E. Braun, V. Meyer, Ber. Dtsch. Chem. Ges. 1888, 21, 1947, https://doi.org/10.1002/cber.188802101376
[8] - E. Braun, Ber. Dtsch. Chem. Ges. 1889, 22, 556, https://doi.org/10.1002/cber.188902201125
[9] - Doug's Lab, YouTube 2015, "Isopropyl Nitrite", https://www.youtube.com/watch?v=bbmpmLjfkkM
[Edited on 18-11-2022 by Diachrynic]
we apologize for the inconvenience
|
|
|
Boffis
International Hazard
Posts: 1867
Registered: 1-5-2011
Member Is Offline
Mood: No Mood
|
|
Diachrynic; nice write-up; its a pity about the yield. I may have a go at this synthesis myself when time permits. By the way what did you product
smell like? Biacetyl monoxime is practically odourless but tetramethyl pyrazine has a strong roast nut-chocolate and to me roast meat smell.
By the way biacetyl monoxime is stable for several years if pure and dry.
I also finally got round to doing a bit more on my dimethyl pyrazine via biacetyl bisulphite complex and ethylenediamine. I steam distilled it and the
oxidised the distillate with NaOH and KMnO4, I may have added too much permanganate but I didn't know how much material was in the distillate. It
didn't smell that strong but it did at least smell about right, earthy and nutty. I hadn't had much success with the ether extraction on the previous
attempt so this time I decided to use Jorre's method of isolation and tried the mercuric chloride solution but only on a small part of the distillate
first after first neutralising it with a few drops of acetic acid. I got a cream coloured ppt which I filtered off, washed and dried to get 0.761g of
material which is equivalent to 0.216g of dimethylpyrazine so I haven't bothered to react it with NaOH to liberate the free pyrazine, it simply isn't
worth the effort.
My experiment with the oxidation of 2,3-dimethyl quinoxaline didn't go very well either. The permanganate oxidation seemed to go OK but when I
evaporated down the light brown filtrate it became dark brown and when acidified nothing crystallised out. More reading up revealed that the acid is
much more soluble than say the triazole dicarboxylic acid I had made before by this procedure but it sounds like the copper salt is easily
precipitated and sparingly soluble. I only used 1.7g of crude starting material on this attempt. When I get time I will try again with pure starting
material and I may also try a hydrogen peroxide oxidation route since permanganate oxidises the methyl groups too.
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Hi Boffis, thanks for kind words and input!
| Quote: Originally posted by Boffis | | I may have a go at this synthesis myself when time permits. By the way what did you product smell like? Biacetyl monoxime is practically odourless but
tetramethyl pyrazine has a strong roast nut-chocolate and to me roast meat smell. |
Hm, that's not a good sign. What I got barely had a smell. It might just be diacetyl monoxime then, it would fit the melting point. Disappointing,
because Wallach was praising how he got such nice and quantitative yields while he claims acidic reduction gave poor yields.
Sad to hear that your attempts have also been of poor success. I don't understand why the literature is so difficult to reproduce...
I have bought some pyrazine as a reference substance, but maybe I will use a small amount to also test the copper(I) complex from the opening post.
So, next I need to test my supposed tetramethylpyrazine, maybe try a different reduction, I need to make 2,3-dimethyl-5,6-dihydropyrazine again and
get the hang of that procedure (clearly both steps need big work and I can't continue trying to do it in one pot), have a look at my manganese(IV)
oxide and maybe see if some other oxidizing agent works better.
Good luck on the 2,3-dimethylquinoxaline oxidation! I assume the product is the tetracarboxylic acid?
we apologize for the inconvenience
|
|
|
Diachrynic
Hazard to Others
Posts: 226
Registered: 23-9-2017
Location: western spiral arm of the galaxy
Member Is Offline
Mood: zenosyne
|
|
Looking for literature on possible side reactions of the ethylene diamine / diacetyl reaction, I came across these reports by Yamaguchi et al.
"Formation of Tricyclic Heterocycles from the Condensation Reaction of 1,2-Diamines with 1,2-Diketones" by T. Yamaguchi, M. Eto, K. Watanabe, N.
Kashige, K. Harano, Chem. Pharm. Bull. 1996, 44, 1977–1979. doi:10.1248/cpb.44.1977
"New compounds derived from dihydropyrazines having DNA strand-breakage activity" by T. Yamaguchi, M. Eto, K. Harano, N. Kashige, K. Watanabe, S. Ito,
Tetrahedron 1999, 55, 675–686. doi:10.1016/S0040-4020(98)01079-5
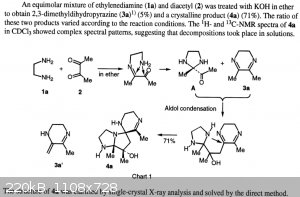
A more detailed description of their procedure can be found in the second publication. Note how low the yield of dihydropyrazine is (5-10%) and how
similar the procedure is to the synthesis of the dihydropyrazine cited in previous posts. This leads me to believe this is probably the mechanism by
which the yield is lost in this procedure. But how to prevent it I'm not sure on.
we apologize for the inconvenience
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
I'm doubtful of the reaction of ammonia with chloroacetone. Aside from the noxious properties of the precursors, the potential for imine formation and
other side reactions is very high.
But butanone easily forms the nitrosated product that Orgsyn calls "dimethyl glyoxal monooxime". Reduction or reductive amination of this under the
right conditions should give 2,3,5,6-tetramethylpyrazine. You seem to be aiming for any pyrazine, and this is certainly one of them.
|
|
|
|









