| Pages:
1
2 |
vanBassum
Hazard to Self

Posts: 66
Registered: 16-4-2019
Member Is Offline
|
|
First yield of KClO3 from my cell
Hello everybody,
I don't know if there is something like a "show your projects" section on this forum but I like to share my first results on a test run I did with my
chlorate cell. I don't have exact numbers yet since this was just a test to see if the materials of the cell would hold up. Especially since I 3d
printed the lid myself and I couln't find any information about the chlorine resistivity of PETG. I just know that it's resistant to a lot of things
so I gave it a go.
My cell is build with a MMO anode and a Ti cathode witch are glued into a lid I printed with my 3D printer. Besides the anodes there are also 2 glass
tubes in the lid. One is closed off and can be used to insert a thermometer while the other is open and used to vent of the gasses. The cell holds
about a liter electrolyte witch consists of a saturated solution of NaCl with a pinch of K2Cr2O7. The cell was run for 4 days straight while the
temperature changed between 60°C to 80°C, due to the changes in ambient temperature. I unfortunately cannot do not know the current it ran at
because I don't have a way to measure this (yet). One of the projects I am currently working on is a device that measures voltage, current and
temperature and logs these values to a SD card over time. I will be using this in the future to measure the cell and calculate the efficiency.
The product you see in the attachment is KClO3 witch is precipitated from the solution by a double displacement reaction between KCl and NaClO3. The
slight yellowish color is due to the K2Cr2O7 witch is still present. I would like to do a crystallization to gain some purity. All the filtrate will
be saved for a future run, a bit of potassium in the cell couldn't hurt but I hope that the solubility won't get to low. Any tips or ideas on this?
Besides, i don't really have a goal for the KClO3.
I used to do some pyro back in the day but KClO3 is something I never used and never going to use in pyro due to safety reasons.

|
|
|
fusso
International Hazard
Posts: 1922
Registered: 23-6-2017
Location: 4 ∥ universes ahead of you
Member Is Offline
|
|
Why not just electrolyse KCl at the beginning to save yourself from the double displacement step?
|
|
|
Heptylene
Hazard to Others
Posts: 319
Registered: 22-10-2016
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by fusso  | | Why not just electrolyse KCl at the beginning to save yourself from the double displacement step? |
I did this with my MMO/Titanium chlorate cell, but found that you have to use very pure KCl to start with. I used 99.3 % pure and ended up with a
slightly beige powder. Recrystallization was necessary to get rid of the color.
|
|
|
woelen
Super Administrator
Posts: 8082
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
If you want a reasonably pure product you have to do a recrystallization anyway. With KClO3 that is much easier than with NaClO3, due to the huge
difference in solubility of KClO3 in cold water, compared to solubility in hot water.
I made KClO3 directly from KCl of decent purity with a pinch of K2Cr2O7, using MMO at the anode and Ti at the cathode. I had the cell in a layer of
water, so that the bottom is cooler than the rest. Crystals of KClO3 form at the bottom. After running the cell, I took out the KClO3 and press-dried
it in a coffee filter, wrapped in paper tissue. Next, I dissolved the KClO3 in as little as possible boiling water and then allowed to cool down
slowly, first to room temperature, then in a freezer to a little below zero (do not let it freeze). Then I decanted the cold water from the crystals
and again press dried them in a coffee filter, wrapped in paper tissue. The result is pure KClO3, good for any chemical experiment (do not use
chlorates for pyro if you value your limbs and eyes!!!) and having a nice white color. If you want it to be really white like snow do a second
recrystallization. Recrystallization only leads to small losses, due to the low solubility of KClO3 in ice cold water.
|
|
|
Simoski
Hazard to Self
Posts: 82
Registered: 24-12-2017
Location: Johannesburg South Africa
Member Is Offline
Mood: No Mood
|
|
Well done vanBassum, God what a victory when you finally get your first crop. The guys are right, electrolysis KCl rather if you want KClO3, it's 5
times easier. And as for PETG 3D printed parts I use them too as the electrode assembly spacers / brackets and what I can tell you is this, they will
last month's immersed in the cell liquor but there is one thing you have to be weary of, crystals, they grow within any space in the PETG part and
slowly tear them apart so print with 100% infill, 0% space.
Again well done
|
|
|
markx
National Hazard
Posts: 650
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Well done! The dichromate additive can be omitted for simplicity sake. It is poisonous, carcinogenic and a strong sensitizer with permanent effects.
The mist from electrolysis can carry it out from the cell, so it tends to spread and contaminate the surroundings.
There are safer alternatives available for the prevention of cathodic reduction:
Attachment: Chlorate und Perchlorate.pdf (2.2MB)
This file has been downloaded 961 times
At page 288 in the abovementioned text one can find the comparative table with different reduction prevention additives for chlorate production.
[Edited on 25-4-2019 by markx]
Exact science is a figment of imagination.......
|
|
|
Heptylene
Hazard to Others
Posts: 319
Registered: 22-10-2016
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by markx | Well done! The dichromate additive can be omitted for simplicity sake. It is poisonous, carcinogenic and a strong sensitizer with permanent effects.
The mist from electrolysis can carry it out from the cell, so it tends to spread and contaminate the surroundings.
There are safer alternatives available for the prevention of cathodic reduction:
At page 288 in the abovementioned text one can find the comparative table with different reduction prevention additives for chlorate production.
[Edited on 25-4-2019 by markx] |
Which book is this from?
|
|
|
woelen
Super Administrator
Posts: 8082
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
I do not agree with markx about the chromate or dichromate addition.
This indeed is not the best stuff to inhale, but it can be kept inside the cell completely. If you vent off the gas from the cell, but put some paper
tissue at the end of the tube, which leads out the gas, then you capture all aerosol. You can also bubble the gas through a solution of NaOH. This
holds back chlorine, but also holds back any aerosol. This is what i did in my setup and I had no issues at all. In the scrubbing solution of NaOH, I
however, did not see any yellow of chromate, so the amount released into the air from the cell is negligible. Your cell must be covered well with a
good cap and the venting tube must be long (several tens of cm at least).
If you leave out the K2CrO4 or K2Cr2O7, then the yield goes down a lot. In my setup, the yield also went down to zero. I still had full electric
current through the cell, but hardly any production of H2 at the cathode. This means that at the cathode I had nearly 100% back-reduction to chloride.
The use of K2Cr2O7 also makes construction of the cell very simple. No membranes needed, just stick your electrodes in the liquid and apply a voltage
over the cell.
The toxicity of K2Cr2O7 also must not be exaggerated. It is not instant death in a bottle. Use common sense when working with it, it indeed is a
sensitizer in some people and it is a carcinogen when inhaled, but some simple measures can be used to avoid exposure. And if you accidently get a
little exposure, then you're not screwed like some people suggest. It is as with smoking cigarettes, a single one does not kill you. Be careful, try
to avoid exposure, and if ou accidently get some on your hands, rinse with water and if you have yellow stain, treat briefly with 2% HCl to which a
little Na2S2O3, Na2SO3 or Na2S2O5 is added. Instant destruction of the hexavalent chromium while no damage is done to your skin.
|
|
|
fusso
International Hazard
Posts: 1922
Registered: 23-6-2017
Location: 4 ∥ universes ahead of you
Member Is Offline
|
|
markx does the file have an english translation?
|
|
|
markx
National Hazard
Posts: 650
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
The text is from "Enzyklopädie der technischen Chemie": Encyclopedia of applied chemistry. Unfortunately I have no english version of it, but I'm
sure the book has been also released in english language. Makes for a great motivation to learn german though 
In my old university chemistry building we had a really ancient and large original set of german literature dating back to the 19th century and even
farther. The treasures that could be picked from there were limitless. Very detailed descriptions of experimental proceedings and conditions. Quite
unique.
Exact science is a figment of imagination.......
|
|
|
vanBassum
Hazard to Self
Posts: 66
Registered: 16-4-2019
Member Is Offline
|
|
Hello everybody,
The ultimate goal would be to build a KClO4 cell. A drawback of KClO4 is the low solubility, therefore I would like to start out with NaCl to create
NaClO3 and oxidize that further to NaClO4. At last a double displacement would leave me with KClO4. I don't expect to do this anytime soon though
since I don't like messing around with soluble lead salts to make my own electrodes and platinum is a bit above the budget right now. Also, I've read
that platinum could be destroyed when the ClO3 contents become to low, so lead oxide would be preferred.
I have given the K2Cr2O7 some thought before use because of its toxicity but I think if everything is contained in the cell with a tube bubbling
through a neutralizing solution of some sort it will be manageable. I am using a gas washing bottle with a fritted disk for this, so I am not to keen
on using hydroxide since that will attack the disk. And those bottles are not cheap... Right now its just filled with water and a bit of sodium
carbonate.
I currently don't own a beaker large enough to do a re-crystallization so I ordered some online but that will take some time to arrive. I also have
thought about using something like a glass coffee pot as a beaker although I don't know if it is suitable. Besides I am going on a vacation in a short
while so the re-crystallization has to wait.
On another note, the PETG and glue holds up quite nicely. Only some discoloration in the plastic is noticeable but its hasn't weakened for as far as I
can tell. I will cut it in half eventually because it needs a small redesign at witch time I'll do a closer inspection. The lid is pressed in the
glass container using a FKM ring to seal it, but it is a bit to small so it doens't seal properly. I included a picture to show the general idea.
Yesterday is started to work on the current amplifier to measure the current and voltage. The principle of operation can be found here: http://tinyurl.com/yyx4db2z I have soldered this to a prototyping PCB but something isn't quite right yet so I'll have to fix that. Ones I'm happy
with it I'll share some photos and source for the arduino so people can recreate it.
@Woelen, Leuk om een andere nederlander te zien, I have used the information on your site when building this cell. It explained everything very
clearly. Besides, you mention K2Cr2O7 is a sensitizer, do you mean that in a health or chemically stability perspective?
Regards,
Bas.

|
|
|
markx
National Hazard
Posts: 650
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Very nice cell construction! If you are not sure about the sealing efficacy of the gaskets and see no heavily detrimental effects upon productivity
without the dichromate, then I would advise to avoid the additive, unless you are conducting the synthesis run outside. In my practice I've not had a
situation where the cell would show such low productivity that additional "boosters" are unavoidable or dearly needed. Even with nonoptimal electrode
sizes and conditions. Woelen mentioned that his cell was of nonagreeable efficacy without the dichromate.....perhaps your set of conditions is similar
and the additives are well justified in that case. It sure does help with raising the current efficiency of the synthesis and it is the industry
standard, but it is nasty stuff with cumulative effects on health. Causes the immune system to act upon it with inflammatory response, as if it was a
foreign disease and to remember that case for future. After that has happened, a very low trace concentration entering your system can provoke an
inflammatory reponse, hence "sensitizer". It is not really an "inhale once and drop dead situation" of course, but just saying. Some people are more
prone to develop this kind of hypersensitivity and some can work with dichromates for decades, not showing any considerable effects. So it is a rather
vague hazard 
Soluble platinum salts (platinum group metal salts in general) are way worse at creating this kind of effect in the human body. As far as I know
hexachloroplatinic acid is the most potent sensitizer that is known and the effects are irreversible once they emerge. I always work with utter care
to avoid this stuff from making contact with my skin or being dispersed into air when I make anodes. It is really scary stuff compared to dichromates
and it fills me with horror to see the techniques that are used by some precious metal refiners who handle coupious amounts of these solutions.
Boiling them down for concentration purposes and conducting reactions that release gases which disperse the solution droplets into air.
But enough moaning about the hazards.....don't wan't to appear like a "total mincer" !
As far as perchlorate synthesis is concerned you can basically take two working routes: Pt based electrosynthesis or the thermal decomposition of
chlorates. Lead dioxide anodes in the amateur setting do not really appear as viable option. The deposition can be conducted rather easily, but
achieving a coating that is durable in perchlorate cell conditions is rare as I've learned from the experience of others and my own experiments. The
coating can seem quite durable in milder conditions e.g. electrolysis of sulfuric acid for ozone generation, which in itself is already rather
agressive as far as anodic conditions go, but the perchlorate cell is a step further (or at least sideways) and seems to make short work of the lead
dioxide coatings. The most I've stretched out of my humble attempts are a few days worth of operation before the anode layers are dispersed into
solution.
If Pt is out of the question because of cost then the thermal decomposition route remains as the option. Actually it is a pretty quick, cheap and
efficient way to process bulk quantities with acceptable yields. The precondition is that your chlorate has to be void of certain metallic
contaminants that act as catalysts upon the decomposition and steer the process towards evolution of oxygen and chloride, preventing any perchlorate
from forming. Manganese and iron can be noted as such detrimental contaminants, but many other metal salts have this property to a smaller or larger
degree.
Once your chlorate cell is operating at expected productivity, the thermal decomposition is a really attractive option for the next stage of
conversion. To be honest, even a moderate sized cell can pile up the chlorate stock at a far greater pace than one can utilize in any reasonable way.
Exact science is a figment of imagination.......
|
|
|
vanBassum
Hazard to Self
Posts: 66
Registered: 16-4-2019
Member Is Offline
|
|
Hello,
The thermal decomposition is also one on the list of things to try although I haven't done any research on that topic yet. As far as I'm aware the
chlorate gets destroyed into chloride and perchlorate witch can than be separated. I do have to do some reading on the topic of what to do and
especially not to do. For instance, what is a suitable crucable? I was always a bit discouraged by the relative low yield of the process, but now my
cell works I have relative cheap access to virtually unlimited chlorate.
About the dichromate, ones I have a good way to measure the efficiency I'll try a run with and without it to compare the differences. That should
yield some valuable information.
|
|
|
phlogiston
International Hazard
Posts: 1381
Registered: 26-4-2008
Location: Neon Thorium Erbium Lanthanum Neodymium Sulphur
Member Is Offline
Mood: pyrophoric
|
|
A porcelain crucible is often suggested.
You'll mainly want to make sure that the melt is free from metallic impurities that can catalyse the decomposition of chlorate to chloride.
You can remove trace amounts of remaining (di)chromate by adding barium chloride (barium dichromate is insoluble around neutral pH).
I recall this method was even used industrially for preparing potassium perchlorate, until better methods were discovered.
PS. Welkom landgenoot
[Edited on 26-4-2019 by phlogiston]
-----
"If a rocket goes up, who cares where it comes down, that's not my concern said Wernher von Braun" - Tom Lehrer |
|
|
vanBassum
Hazard to Self
Posts: 66
Registered: 16-4-2019
Member Is Offline
|
|
I got around doing a re crystallization. After witch I dissolved everything again and used a bit of BaCl2 to get rid of the chrome. This was done by a
hot filtration, I could clearly see some greenish particles in the filter. After everything was crystallized out I did a small washing step and put
everything on a dish to dry. (I do have a lab microwave / grill witch i got for free from a fried.)
Right now I am trying the decomposition. My hotplate is fortunately hot enough to melt everything in a beaker and keep a fairly steady temperature.
Its liquid but not bubbling. Only a few very small bubbles are floating around. A bit like coke in a glass. I some very small white precipitate
floating around in there. This is presumably the percholate or chloride.
I have some methylene blue but it didn't turn purple in a solution known to be KClO4. Any suggestions?
[Edited on 1-5-2019 by vanBassum]
[Edited on 1-5-2019 by vanBassum]
|
|
|
markx
National Hazard
Posts: 650
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
Methylene blue is not very sensitive towards mediocre perchlorate concentrations (as in the level created by the low soluble KClO4). If I percipitate
the perchlorate from cell liqour sample with KCl and test with methylene blue I also get a barely noticeable reaction, so it seems a normal course of
events.
Exact science is a figment of imagination.......
|
|
|
yobbo II
National Hazard
Posts: 777
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
Methylene blue is very sensitive towards perchlorate. Very small concentrations of perk turn the stuff purple.
|
|
|
markx
National Hazard
Posts: 650
Registered: 7-8-2003
Location: Northern kingdom
Member Is Offline
Mood: Very Jolly
|
|
| Quote: Originally posted by yobbo II | | Methylene blue is very sensitive towards perchlorate. Very small concentrations of perk turn the stuff purple. |
Hmm....my limited experience so far has shown it to be quite dull towards low concentrations of potassium perchlorate. Perhaps the the presence of
K+ plays a role in this phenomenon or the indicator substance sample that I use is somehow "special"?
With sodium perchlorate solutions the reaction is sharp, permanent and very evident, but testing on the potassium salt I tend to get a slow, barely
noticeable and temporary color change.
Exact science is a figment of imagination.......
|
|
|
yobbo II
National Hazard
Posts: 777
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
I obtained a concentrated solution of sodium chloride and a conc. solution of K chloride and added the same amount of perchlorate to each. About 3
grams per litre I reckon. The methylene blue is less sensitive to the solution containing the K. It would not really show up this concentration in the
K solution but easily showed up the perk. in the Na solution. So you are right!
Yob
|
|
|
yobbo II
National Hazard
Posts: 777
Registered: 28-3-2016
Member Is Offline
Mood: No Mood
|
|
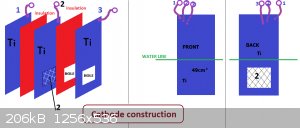
Put together some cells to have a look at the current distribution on a cathode together with the cathodic reduction 'thing' (wanted products being
converted back to starting products which we do not want) .
I have read that dichromate effectively raised the current density by forming a film on the cathode which reduces/eliminates the cathodic reduction
and at the same time not giving a higher voltage accross the cell [more power usage, a bad thing] Perhaps this is too simplistic of an explanation.
It is stated elsewhere that the film stops hypochlorite from touching the cathode and allowing other stuff to pass through thus reduction of the
hypochlorite cannot take place.
I started with a small MMO anode that was about 14 cm squared on each side.
The cathode (Ti) is large (too large) and is approx. 49cm^2 on each side.
The first cell with the large cathode contained about 1.75 litres and was placed on a hot plate
to warm it up as the current running was unable to get the temperature very high.
The cell was let run for about 14 hours to allow hypochlorite to build up before any measurements were taken.
Current going to/from the cathode was measure using four 0.015 Ohm resistors. One resistor measured the total current with
the other three resistors measuring the way the current split between cathode front, outside back and middle back.
With the temperture at 29C and total cathode current at 5.33 Amps the front cathode current was 4.06 Amps, middle back was
0.37 Amps and the outside back was 0.87 Amps. When temperture was 58C the currents were 3.83, 0.42 and 1.03 Amps. The
higher the temperature the more conductive the solution is, which gives more current on the back side of the cathode.
The larger graphite anode (of similar dimensions to the cathode) give the following at 60C with anode current at 5.33 Amps. 3.54 Amps on front, 0.57
middle back
and 1.2 Amps on back outside of cathode.
As expected there is much lower current density on the back side of the cathode.
Gas measurements were taken using an 500ml volumetric flask filled to its mark with water and inverted in a bowl of water.
The same cathode/anode current (5.33 Amps) was used for all measurements for the first cell.
It took 18.2 minutes to fill (500ml gas) with all parts of cathode running current at 72C.
With only the front of the cathode connected, (same anode current) the time taken to fill 500ml was 15.66 minutes (more gas produced).
Four grams of Na2Cr2O7:2H2O (Sodium Dichromate) was added to the cell. The temperature of the cell fell about 7C from 72 to 65C
(without adjusting the hot plate setting of course) as time went by.
Running current to both sides of the cathode, the time taken to fill 500ml with gas was 12.66 minutes.
Running current to the front of the cathode only, the time to fill 500ml was the same (12.66).
A few hours later the time to fill 500ml was 12 minutes. More gas is being produced when the Na Dichromate is added.
Another cell was set up, this time with a much smaller cahtode (Ti) with the back side of the cathode permanently covered with a plastic
covering so only current went to the side facing the MMO anode. The anode was the same as the first cell and the current was set at
three different values. Working surface area of the cathode was approx. 7cm^2. The hot plate was not used to heat the cell.
Nothing can be read into the different temperature reading as not enough time was given to the cell to stabalize at a given temperature
as currents were varied.
Green cell.
The time taken to produce 500ml of gas at 5.33 Amps was 13.13 minutes. (5.35V accross cell, about 35C)
The time taken to produce 500ml of gas at 2.66 Amps was 25.70 minutes. (4.16V accross cell, about 25C)
The time taken to produce 500ml of gas at 1.33 Amps was 48.00 minutes. (3.90V accross cell, about 18C)
Four grams of Na Dichromate was added to the cell.
The time taken to produce 500ml of gas at 5.33 Amps was 12.0 minutes. (5.26V accross cell, about 30C) [3 hours later it was 12.75 minutes]
The time taken to produce 500ml of gas at 2.66 Amps was 25.65 minutes. (4.57V accross cell, about 21C)
The time taken to produce 500ml of gas at 1.33 Amps was 51.66 minutes. (3.6V accross cell, about 20C)
The is still a time difference between Dichromate and no Dichromate, green and non-green! in favour of the Dichromate but
it is not so large with the smaller cathode with the much higher current density.
(The green cell is actually doing better for the low current run).
The voltage accross the cell will be higher
when a high current density is run on the cathode.
There is quite alot of gas coming from the anode, Oxygen I presume. Dichromate also supresses O generation but it is difficult to
tell the difference between the two situations by looking at the gas coming off the anode. The reduction of O generation
(assuming it is happening) will reduce the 'gas-measured-score' that is being attributed to the addition of Dichromate.
I have read somewhere that it takes a few hours for the anti-reduction film to build up properly on the cathode but is seemed
sudden enough with these cells.
The pH of this cell when measured at 7.7 when gas measurements were ended.
@ Woelen You came up with a figure of 200mA (and above) per square cm as a threashold for cathodic reduction not being a problem. Just wondering where
or how you came up with that figure?
I hope to make two similar K cells, put them in series. One with a small cathode, other with normal cathode + dichromate and see if there is much of a
difference in KClO3 output. I have always thought the the dichromate give something like a 3 to 6 % current efficiency boost to commercial cells. But
I am only guessing. There are other reasons for adding it to cells. It's a buffer, it also protects steel cathodes and other steel components
(everything nowadays is probably Ti and teflon) and it also lowers oxygen generation at the anode.
Stay green!!!
Yob
[Edited on 19-5-2020 by yobbo II]
|
|
|
mysteriusbhoice
Hazard to Others
Posts: 477
Registered: 27-1-2016
Member Is Offline
Mood: Became chemistry catboy Vtuber Nyaa
|
|
in my case Ca(Cl)2 messes with my runtimes too
it causes deposits on my Ti cathode and overtime it gets covered in a white crust until its gone from the cell.
If I do clean the deposits every day or few hours the runtime gets shorter and yield goes up!!!??
It turned out my Ph stayed at 7-8.5 (I have a pH meter) as long as the calcium chloride was in the cell until it got all coverted to Ca(OH)2 idk if
overtime this may damage the anode in some unforseen way but it is present in some quantities in sea salts.
I usually run till 60-70% conversion so I dont end up making perchlorate and killing my MMO due to these unexpected run time changes and even with
predicted 54% efficiency I end up getting something like 80% conversion which is way off!! and it means Ca(Cl)2 seems to be a self regulating pH
buffer until its consumed however it does form bad crust on cathode while it converts and there are no chromates in my cell.
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 198
Registered: 12-7-2018
Member Is Offline
|
|
What i read you can have an efficience of 87% with CaCl2. The CaOH forms an Diaphragma on the Cathode and there is an less reduction from the
hydrogen. I have tried it too but when you dont use an magnetic stirrer it dosent work or it doesnt work for me. There where only two layers on the
top i had an PH of 6 and on the Ground an PH of 12.
[Edited on 21-5-2020 by Alkoholvergiftung]
|
|
|
mysteriusbhoice
Hazard to Others
Posts: 477
Registered: 27-1-2016
Member Is Offline
Mood: Became chemistry catboy Vtuber Nyaa
|
|
| Quote: Originally posted by Alkoholvergiftung | | What i read you can have an efficience of 87% with CaCl2. The CaOH forms an Diaphragma on the Cathode and there is an less reduction from the
hydrogen. I have tried i too but when you dont use an magnetic stirrer it dosent work or it doesnt work for me. There where only two layern on the top
i had an PH of 6 and on the Ground an PH of 12. |
I read that depending on how much turbulence is caused by the microbubbles and depending on the shape of your cell it can act like a plug flow or
mixed flow reaction vessel.
I put about 25 amps through my cell and the walls are very close creating lots of turbulence and hence perhaps thats good enough stirring to allow it
to work because.
I was expecting 1kg of NaClO3 after 4-5 days didnt really count but instead got 1.6kg with inputted 1055 grams of NaCl which means about 89 % CE since
the chlorate was a bit damp it could be more like 1.5kg which is on the ballpark of 87% efficiency.
All i did was use natural sea salt MMO electrode 2x and a single cathode.
reducing the surface area of the cathode is what I went for to promote anodic oxidation.
current density of 88ma/cm^2.
Forget chromates
CaCl2 FTW
and if you run sea salt it will contain large amount of CaCl2 depending on your source.
I bought the cheapest edible salt due to low cost and btw it tastes horribly bitter and soapy prolly due to high CaCl2 quantity.
It said at the back naturally extracted sea salt or some crap and due to how fine the salt was and it being really damp upon opening not a free
flowing powder i think they just boiled some brine and called it good.
[Edited on 20-5-2020 by mysteriusbhoice]
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 198
Registered: 12-7-2018
Member Is Offline
|
|
Mysteriusbhoice, is your cell an open design, withour an lid? If there is no mixing like in my case the most chlorine get lost.When i work in my
Standard cell with NaCl and KCl and an Elektrode distance of 5mm i can work without lid and there is no chlorine smell.
|
|
|
mysteriusbhoice
Hazard to Others
Posts: 477
Registered: 27-1-2016
Member Is Offline
Mood: Became chemistry catboy Vtuber Nyaa
|
|
| Quote: Originally posted by Alkoholvergiftung | | Mysteriusbhoice, is your cell an open design, withour an lid? If there is no mixing like in my case the most chlorine get lost.When i work in my
Standard cell with NaCl and KCl and an Elektrode distance of 5mm i can work without lid and there is no chlorine smell. |
My cell is a closed lid with gasket and temps are around 60-65 celsius and capacity is 4L and theres a bubbler leading to the outside.
I dont add KCl since right now im testing out GSLD electrodes to convert the product from that cell into perchlorate.
The thing with KCl is that the insolubility of KClO3 will cause the current to drop over time.
As for my cell even after the end of the run the smell of chlorine was so noxious!! even after full run time chlorine smell was so prevalent I had to
open it outside.... I couldnt even open the cell without getting a strong wiff of Chlorine and hence the bubbler being present.
I really think CaCl2 is amazing but dont use it with potassium salts because the insoluble Ca(OH)2 will force you to recrystalize that stuff.
When I use that nezo brand salt which is 71%KCl and 29% NaCl by wt my CE is usually only 50% region.
When i used that fine raw sea salt which has CaCl2 my CE goes crazy without chromates or shit.
I also read a paper that soluble permanganates increase CE also.
The toxicity of permanganate is very low compared to dichromate because it easily reduces to MnO2.
https://sci-hub.tw/https://www.sciencedirect.com/science/art...
My cell is really old and constructed approx 2 years ago
[Edited on 21-5-2020 by mysteriusbhoice]
[Edited on 21-5-2020 by mysteriusbhoice]
 
|
|
|
| Pages:
1
2 |









