Klute
International Hazard

Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Attempted synthesis of thiols by reduction of Bunte salts (alkylthiosulfates)
Last week I attempted the synthesis of 2,5-dihydroxythiophenol by Zn/HCl reduction of the Bunte salt formed by reacting benzoquinone and sodium
thiosulfate.
The two steps are best performed one-pot, as isolation of the Bunte salt is tedious (evaporation of a large volume of conc. salt solution) and
wastefull. The excess thiosulfate doesn't cause a problem during the reduction, nor does the acetic acid.
The first reaction proceeded very smoothly, and was very easy to perform. The reduction was another story.
Reduction of Bunte salts generate copious amounts of H2S. The original work by W. Alcalay (Translation of the original Helv. Chim. Acta
article joined) suggests gradually adding the zinc in small portions after adding the acid. I didn't feel confident in opening such a setup during the
reduction, and was right to. So i decided on reversing the addition, adding all the zinc at the beggining, and then gradaully adding the acid.
Magnetic stirring was very difficult, and the addition took many hours to complete without producing too much foam. Apart from the that, the reaction
seemed to work well. But when I started the workup the next day, things went bad. During the extraction of sludge, in the agrage on a very windy day
with all doors opened, several ventilators around the bench, and a gas mask on, I got intoxicated with the H2S still emmiting from the reaction
medium. A description of the effects and the administration of sodium nitrite as an antidote can be read here.
Nevertheless, I decided on posting the details of the reaction.
I strongly advise anyone willing to try this reaction in amateur settings to reconsider. Other, longer but safer reaction paths can lead to
thiophenols. H2S doesn't give you a second chance, I was very lucky of surviving and not having any serious remaining/delayed health effects.
Synthesis of 2,5-dihydroxythiophenol
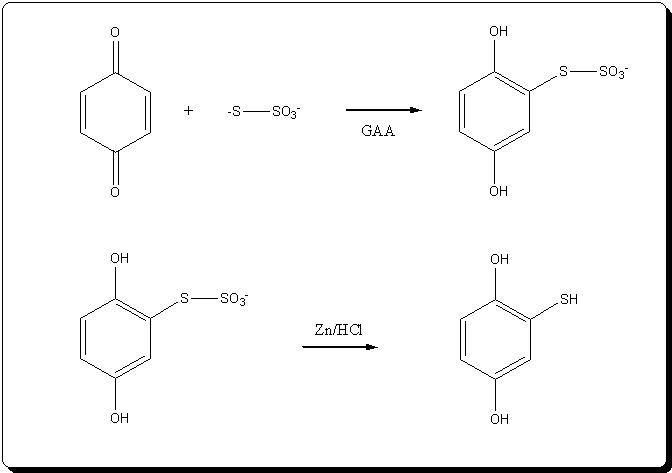
Step 1: Formation of the Bunte salt
In a 500mL 3-neck RBF, equipped with a condenser, a addition funnel, a thermometer and magnetic stirring, 75.0g (302.2 mmol, 1.5eq) of sodium
thiosulfate pentahydrate are dissolved in 100mL H2O. the gradual addition of the crystals form a cold milky solution, the dissolution being
endothermic.

21.5g (198.9 mmol) of benzoquinone (Note 1) are dissolved in 100mL GAA with warming. A dark red solution is obtained, and 1/4 charged in the
addition funnel (Note2).

The RBF is cooled in a ice bath, an dthe quinone slowly dripped in with steady stirring. As soon as hte solution touches the thiosulfate solution, it
formed a dark red colour, and a viscous oil-like material that slowly dissolves. The solution cristallizes as it touches the cold sides of the flask
(Note3). The formed crystals are gradually washed down, turning red, then black on contact with the thiosulfate. The milky solution quickly
clears up, and gradually turns rusty brown/red. the temperature is kept at all times under 10°C.




The addition lasts 1 hour, and the flask is stirred for 2 more hours as the ice bath melts.
Step 2 : Reduction of the Bunte salt
The solution obtained in the previous step is transfered into a 1L 3-neck RBF, equipped with: a condenser attached to a 30% wash bottle, followed by a
12% NaOCl wash bottle, a 250mL addition funnel, a thermometer and magnetic stirring.


To the stirred solution, 50g of Zn dust are added in one portion. There is some gas evolution, and a exothermic reaction sets in. The temperature
climbs over 60°C, so a ice bath is applied until the reaction calms down. Once gas evolution slackens ( a few minutes), the rest of the zinc is added
in two portions. A total of 100g (1.53 mol) of zinc dust are added. The temeprature is 45°C.


250mL of 31% HCl are charged in the addition funnel, and slowly dripped in, with the fastest stirring the stir bar can handle without jumping. There
is a vigorous gas evolution, and foam starts to form on the surface of the reaction medium. Magnetic stirring becomes very diffcult and inefficient,
so the flask is periodically shaken by hand to break up the foam/crust forming on the surface. A slowly drip is maintained (1drop/1-2sec). The
temperature, after climbing up to 60°C, gradaully comes down to 40°C, at which point the flask is briefly heated up to 60°C with a heating mantle.
At one point the addition funnel is recharged with 150mL 31% HCl, and the H2S smell is detected. The setup wasn't opened for more than 2 sec. Near
the end of the addition, the foaming calms down, and mag stirring is possible again. When the gas evolution diminishes significantly, the flask is
heated to 50°C in a oil bath, giving off a constant flow of bubbles.




A dark gray fluid suspension/sludge is obtained. It is left to stir at RT over night, no more ags evolution noted.
The NaOH wash bottle started turning yellow/orange during the H2S evolution. A white solid (sodium (poly)sulfides) started forming on the bottom of
the immersed tube, and clogged it at two occasions, making a stopper pop and release H2S. It was quickly disconnected, and replaced by the NaOCl wash
bottle, which had not changed apperance since the begging of the reduction. It immediatly started turning cloudy and sulfur particlues were seen
appearing. Each wash bottles heated up when being the first connected during the reduction (up to 40-50°C).


(the disloged white solid can be seen on the bottom, after the first clogging up)


(After stirring overnight)
The next morning, with all the precautions described earlier, and never staying in the garage for more than a couple minutes, breathing fresh air
outside for 5 min before going back in, the setup was disassembled, and the sludge transfered to a seperating funnel. 100mL of DCM were added, but
formed a unseperable emulsion, so 100mL toluene were added, and seperated nicely after extraction. The xtractions were repeated with 2x100mL toluene,
the layers been left to sperate for at least 10min each time. At the third extraction, I started feeling sick.
My hands started shaking strongly, I was about to faint, my vision got blurred a bit and my heart was racing. The rest of that story is in the
dedicated thread...
I must insist that I only smelled the rotten eggs smell a couple of times, when getting out o fthe garage and taking the ags mask off. I'm sure that
the gask mask wasn't that efficient, and that my sense of smell was quickly nubbed up. Never underestimate the toxicity of this compound.
After having recovered (several hours), I neutralized all the glassware with conc. hypochlorite solution, and emptyied the sludge in a jerrican
containing a Ca(OCl)2 suspension. The extracts were saved, washed with 2*100mL brine to remove dissolved H2S, and dried over Na2SO4. The water-clear
extracts still smelled of H2S, so a 30% NaOH wash bottle
was connected to the vacuum inlet of a fractionnal disn setup. The DCM was removed at atm pressure, and washed with 30% NaOH solution as soon as
distd. There was some gas evolution at the begging of the distn, as air was chased out of the setup, an dit gradually stopped as most of the DCM was
distd.
The toluene was distd at 60-70°C under slight vacuum, no H2S smell was noticeable from the water aspirator station (containg sodium carbonate).
Once most of the solvent was removed, a clear, very slightly yellow residu was obtained, still containing toluene. 50mL of pet etehr were added, and
the flask place din the freezer. A insignificant amount of white crystasl appeared (<100mg).

Notes:
Note1: Made from hydroquinone with H2O2 and catalytic I2 in IPA
Note2: Not all the solution was charged to avoid cristn of the quinone in the funnel; it was periodically warmed up with a hair dryer.
Note3: This is best avoided by attaching the addition funnel to a vertical neck, or incling the setup in such a way that the dripping solution touches
directly the thiosulfate solution.
Comments
The major problem was using toluene as extarction solvent. The product is said to be easily recrysatllized in benzene, so I guess it isn't very
soluble in toluene. Unfortunaly, I had no opened bottles of ether, DCM formed a emulsion, and ethyl acetate would have hydrolyzed in a few seconds in
such a acidic environment.
The reduction is said to proceed in 1-2h when adding the zinc to the acid/substrate, maybe the long reaction time degarded the product. But apart from
a solid additon funnel, there are no means of safely introducing zn dust gradually during sucha reaction. Even under a good fume hood, i would
consider it as risky.
So although the reaction looks easy enough on paper, and when described in a >60 years old publication, the adequate conditions are far from
evident for a home-chemist.
There is another way of obtaining thiols from Bunte salts, that is basic or acidic hydrolysis. But according to H. DISTLER ("The Chemistry of Bunte
Salts", Angew. Chem. internat. Edit. 1 Vol. 6 (1967) J No. 6), basic hydrolysis of Bunte salt obtained by addtion of a thio compound on a
double bond simply regenerates the satrting unsaturated compound (here benzoquinone) and the thio compound (thiosulfate). But acidic hydrolysis is
said to work:
| Quote: | From "The Chemistry of Bunte Salts"
f) Reduction of Bunte Salts
The reduction of S-alkylthiosulfates with conventional
agents, such as sodium amalgam1771, zinc and
acid [78,89,93,94,1381, or alkaline sodium arsenite solution17.81
leads to thiols. However, this method is of
little importance for the preparation of thiols, which
can be obtained much more elegantly by cleavage of
S-alkylthiosulfates with acid. |
However, a major side-reaction occures with non-volatil thiols, as they react with remaining S-thiosulfates to form disulfides as soon as formed.
There are much easier and less wastefull means of obtaining disulfides.
There could be a way of avoiding H2S evolution: Bunte salts are readibly converted to sulfonyl chlorides by chlorination in cold aq. conditions
(T.B. Johnson, unknown journal, 25 (1939) 448-452): I.B. Doughlas, T.B. Johnson, JACS, 60 (1938) 1486-89; I'm guessing hypochlorite
solution could work here. And reduction of sulfonyl chlorides with Zn/HCl yields thiols without H2S evolution.
But on the other hand hydroquinone is readibly ring-chlorinated whene exposed the chlorine in acidic conditions (US 2,748,173), so some ring
chlorination could be possible. Oxydation to the corresponding quinone could be possible too, but that would be reduced back to the hydroquinone
during reduction.
Obviously, larger amount sof zinc dust would be needed, making workup somewhat tedious. And I haven't found any example of arylthiosulfates been
oxidized to arylsulfonyl chlorides, only alkyl and benzyl. There could be a reason to this.
So, although this could be a interesting path, it is also a uncertain one, and ideally it would require characterisation of the intermediates with NMR
or at least IR. TLC could give some insight towards the amount of by-products, but not their nature or if the reaction was succesfull or not.
SO if anyone can find further information on chlorination of arylthiosulfates to the sulfonyl chlorides, this would be very usefull fot the synthesis
of thiophenols, but also TsCl and other arylsulfonyl chlorides and their esters.
Attachment: Translation_mercaptobenzoquinones.doc (84kB)
This file has been downloaded 1435 times
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
not_important
International Hazard
Posts: 3873
Registered: 21-7-2006
Member Is Offline
Mood: No Mood
|
|
A way to add solids without opening the system is to place the solid in a small flask and then attach that to the reaction vessel with either an angle
adapter or a section of large flexible tubing.
With the angle adapter you rotate it around the joint with the reaction vessel and tap gently until some of the solid slides out of the small flask
through the adapter.
With the flex tubing you just lift and tilt the small flask until you can tap a bit of solid into the reaction vessel; I think this way is a bit
easier.
Nothing on the arylthiosulfate to sulfonyl chloride path. I too would worry about the hydroquinone being attacked in some way during the conversion.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you for your comments.
Stoechiometric_Steve also suggested to me the use a double-male angle, I will ask to my glassblower how much such a piece of glassware would cost,
even though I will not be trying this reaction again, it can be usefull.
I guess the only way of seeing if the hydroquinone is modified is to try out, and compare with a sample of the corresponding benzoquinone.
I need to rectify something: TsCl can not be obtained as easily, as there is no way of making an arylthiosulfate from an arylhalide and thiosulfate. I
guess that's why there is only example of alkylthiosulfates chlorination in the litterature.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
LSD25
Hazard to Others
Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Klute, I was reading this the other day
http://www.kok.chembio.ntnu.no/chemtalk/Artikler/Schlenk%20t...
On page 7 they show a transfer of a solid from a schlenk type vessel to another schlenk type vessel.
Here is a google book I found today
http://tinyurl.com/6bqfom
Surely that little bent test tube looking thing couldn't possibly be too expensive to have made?
[Edited on 15-5-2008 by LSD25]
Whhhoooppps, that sure didn't work
|
|
|
|








