Tsjerk
International Hazard

Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
synthesis pathway baclofen
I was looking for a new compound to synthesize and my eye fell on baclofen. There are some nice reactions I didn't do before.
The article that caught my attention was the one attached... Well, after stumbling on some strangely formulated sentences (in the discussion), I
started looking at the described reactions.
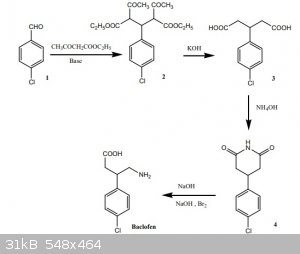
First I wanted to figure out the mechanism of the first reaction and I realized the first step is not a Claisen condensation as claimed in the
discussion, but a sequence of two different reactions, namely the Knoevenagel condensation followed by a Michael reaction (I think). Is this possible?
With piperidine as described in the article this shouldn't be a problem (the condensation reaction forming water), but an alkoxide as discussed in the
discussion would be destroyed by the water. Or is hydroxide strong enough as a catalyst? I have sodium metal, I have sodium hydroxide, but no
piperidine or pyridine.
What surprised me most was the synthesis of the glutarimide ring, directly from the glutaric acid with aqueous ammonia. Is this possible? If not no
problem, then I will do an urea/microwave melt as described below, but a direct glutarimide ring with just ammonia would be easier. It could also be
done via dehydration to the phthalate anhydride with acetic anhydride, but that I don't have.
The Hofmann rearrangement I will do with hypochlorite.
urea / microwave glutarimide synthesis
Paper:
Attachment: ICC_Volume 4_Issue Issue 2, pp. 133-235_Pages 142-145.pdf (84kB)
This file has been downloaded 834 times
Edit: I now realize that 1,1-ish mole of sodium (about 25 gram) as sodium ethoxide, would be enough to drive about 380 grams of reactants to form
product 2 (if 100% yield), not as a catalyst but as reactant. Which doesn't sound too bad.
[Edited on 16-11-2018 by Tsjerk]
|
|
|
clearly_not_atara
International Hazard
Posts: 2826
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
I'm a bit confused: I'd expect to see a dialkyl malonate used in the first step, but acetoacetate is used - and then the product is deacetylated
faster than it's decarboxylated?!
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I understand your confusion... I didn't look at the mechanism of the second step yet.
I'm less good at this than I would like to be, but do you mean that with acetoacetate, and decarboxylation being faster than deacetylation , this
reaction would lead to a ketone instead of a carboxylic acid? As the reaction as proposed in the paper would only go with a diacid?
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
Another route to baclofen:
1.http://revroum.lew.ro/wp-content/uploads/2009/RRCh_11_2009/A...
2.michael addition using excess base
3.reduction
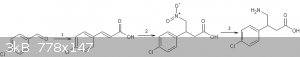
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Looks very doable CuReUS, but I don't have nitromethane anymore and that is one of the few chemicals I can't buy... I finished my last bit I bought
OTC 12 years ago. The route suggested I choose because I have almost all chemicals.
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I'm now thinking about doing the first step with diethyl malonate, as the second step might work with the ethyl acetoacetate phenyl compound, but if
that would work, it will most likely also work with diethyl malonate.
The third step would be performed with urea in a microwave, as I have little confidence in the glutarimide ring formation with aqueous ammonia. The
Hofmann rearrangement should work as predicted.
I still like the predicted reaction scheme as there is no need for large volumes of solvents and no need for distillation as all intermediates can be
isolated by filtration and can be purified by recrystallization.
[Edited on 18-11-2018 by Tsjerk]
|
|
|
Sigmatropic
Hazard to Others
Posts: 307
Registered: 29-1-2017
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by clearly_not_atara  | | I'm a bit confused: I'd expect to see a dialkyl malonate used in the first step, but acetoacetate is used - and then the product is deacetylated
faster than it's decarboxylated?! |
I agree that it is unusual but the decarboxylation of a malonate ester is a two step procedure where the decarboxylation happens under acidic
conditions. The deacetylation of acetoacetate esters occurs at the ketone, entirely under basic conditions. See pages 475-476 in Vogel's practical
organic chemistry.
If you would subject the acetoacetate ester to the two step procedure you would end up with the wrong product. Vogel mentions better yields are
obtained with malonate esters which may or may not be true for this baclofen intermediate.
|
|
|
Assured Fish
Hazard to Others
Posts: 319
Registered: 31-8-2015
Location: Noo Z Land
Member Is Offline
Mood: Misanthropic
|
|
The formation if the glutarimide ring should work alright with ammonia, similar approaches are used to prepare other diacetylamides with ammonia.
http://www.prepchem.com/synthesis-of-succinimide/
https://www.youtube.com/watch?v=Ir3rmvNAgh8
The prepchem example is rather crude but from what it looks like stoichiometrically, both acids are neutralized to their respective ammonium salts
indicating that a loss of ammonia is required during the main dehydration part of the reaction.
Now as for the rest of that reference from the Iranian chemical communications.
That work doesnt look origional to me, the first clue was where they stated using ethyl acetoacetate when in their pathway drawing they clearly used
methyl acetoacetate.
I did a little digging around and found another reference from a paper from what looks like an indian student from 2014.
Attachment: IJIPSRMN-27.pdf (1.1MB)
This file has been downloaded 754 times
Following her references around yielded little results except this 2010 paper that didnt actually have anything about that particular scheme in it.
https://sci-hub.tw/https://www.researchgate.net/publication/...
Anyway my point is, this is not an original scheme, its clear it was netted from elsewhere but precisely where i am having a hard time determining.
Sufficiently advanced science is indistinguishable from madness.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Tsjerk | | I'm now thinking about doing the first step with diethyl malonate, as the second step might work with the ethyl acetoacetate phenyl compound, but if
that would work, it will most likely also work with diethyl malonate. |
I dont understand.Why don't you want
to use acetoacetate when you have the ref for it ? (although all the reactions I have seen that use acetoacetate give ketone rather than carboxylic
acid after base/heat treatment,like atara said above)
| Quote: | | The third step would be performed with urea in a microwave, as I have little confidence in the glutarimide ring formation with aqueous ammonia. The
Hofmann rearrangement should work as predicted. |
actually,you could get baclofen in 1 step from the diacid by
doing a schimdt reaction using 1 mole of azide
[Edited on 19-11-2018 by CuReUS]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
| Quote: Originally posted by CuReUS | I dont understand.Why don't you want to use acetoacetate when you have the ref for it ? (although all the reactions I have seen that use acetoacetate
give ketone rather than carboxylic acid after base/heat treatment,like atara said above)
| Quote: | | The third step would be performed with urea in a microwave, as I have little confidence in the glutarimide ring formation with aqueous ammonia. The
Hofmann rearrangement should work as predicted. |
actually,you could get baclofen in 1 step from the diacid by
doing a schimdt reaction using 1 mole of azide
[Edited on 19-11-2018 by CuReUS] |
I was a bit doubtful about the reference, and after Atara's comment I wanted to go for safe. But I like all your reactions! when I have time I will
have a better look at them... too bad I actually have to work during working hours nowadays. I like your azide idea, I can't find references about it
though, do you have one? Or a bit more about the mechanism?
[Edited on 19-11-2018 by Tsjerk]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
| Quote: Originally posted by CuReUS | actually,you could get baclofen in 1 step from the diacid by doing a schimdt reaction using 1 mole of azide
[Edited on 19-11-2018 by CuReUS] |
How would I separate between non, -mono and di reacted product? I'm pretty sure it will become a mixture of all.
@Assured Fish: Nice digging, I found the Indian mechanism before via Google, it was a figure on Wiki, but not linked to any Wiki page... This
mechanism is doing a dehydration with acetic anhydride, then doing a rearrangement with a nitrogen source. I was guessing urea/glutamic acid microwave
would be easier than the two step, but as Assured Fish says an aqueous ammonia would also work, which is far easier.
|
|
|
clearly_not_atara
International Hazard
Posts: 2826
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Based on what Sigmatropic found in Vogel's, I think that you should give the standard lit route a chance. I find it confusing that
acetoacetate was preferred for this preparation; nevertheless, the double-addition of two Michael acceptors to a carbonyl group is not a common
transformation, so it may work better with some reagents than others for unknown reasons.
The cyclic imide formation and subsequent Hofmann degradation seems like a perfectly workable process, and I see no reason to replace this with the
Schmidt variant of the Curtius rearrangement. The formation of a cyclic intermediate seems like it would have the desired effect on selectivity that
you mention. However, there are a few things we might consider:
- When N-bromosuccinimide is subject to the action of alkali, the ring cleaves to a succinate mono-N-bromo-amide which undergoes Hofmann degradation
as expected. Since the Hofmann degradation is a notoriously fidgety reaction, it might be better performed stepwise by pre-forming the N-bromoimide.
This can be accomplished by dissolving the imide as its sodium salt in water that is not too alkaline, and then adding bromine, which should
precipitate the bromo-imide. The precipitated bromo-imide can then be subject to concentrated alkali to obtain baclofen. This should avoid
over-halogenation.
- It may be possible to convert the diacid to a cyclic anhydride, similar to the formation of succinic anhydride from succinic acid on heating. If
this cyclic anhydride can be formed, it should react selectively with sodium azide or another source of N3- to form the diacid
mono-azide. This will then undergo a Curtius rearrangement targeting exactly one carboxylate group, yielding baclofen with high selectivity. A major
advantage of this technique is that there is no need for the use of hydrazoic acid or any activated source of azide. However, contacting the anhydride
with sodium azide may not be trivial. I do not know if there is a standard way to solubilize N3- in an aprotic solvent. The
Curtius rearrangement is generally cleaner and higher-yielding than the Hofmann degradation, but the most tempting approach to this rearrangement
depends on the generation of the cyclic anhydride (3-(p-chlorophenyl)-glutaric anhydride) which I cannot guarantee.
[Edited on 19-11-2018 by clearly_not_atara]
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
Acetoacetate makes a fine acetyl synthon. The first step is essentially the Hantzsch Dihydropyridine Synthesis without any amine to close the pyridine
ring.
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Tsjerk |
How would I separate between non, -mono and di reacted product? I'm pretty sure it will become a mixture of all. |
if you use exactly 1 mole of azide,dripping it slowly into a pot of diacid,you will have mostly the mono reacted product.As for
separation,you could use the method in the paper you linked above.
more about schmidt -https://en.wikipedia.org/wiki/Schmidt_reaction | Quote: Originally posted by clearly_not_atara |
The cyclic imide formation and subsequent Hofmann degradation seems like a perfectly workable process, and I see no reason to replace this with the
Schmidt |
The only reason I suggested the schmidt was to do the reaction in 1 step in 1 pot rather than going
about a 2 step route.Also the hoffman reaction is messy and low yielding
[Edited on 20-11-2018 by CuReUS]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Why do you think so? This is the first hit I get when I google hypochlorite / glutarimide / Hofmann and yield.
| Quote: | | ....with a small amount, and then washed with isopropanol, and finally with isopropanol-water (1: 1) re-crystallization to obtain a 3-isobutyl-γ-
amino butyric acid 72.0g (76.6% yield), mp 168- 169 ° C. |
source
Edit: Also HN3 makes me a bit nervous, my fumehood fan isn't what it used to be.
[Edited on 20-11-2018 by Tsjerk]
|
|
|
CuReUS
National Hazard
Posts: 928
Registered: 9-9-2014
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Tsjerk |
Why do you think so? This is the first hit I get when I google hypochlorite / glutarimide / Hofmann and yield. |
https://erowid.org/archive/rhodium/chemistry/methylamine.ace... - 55% yield
Also , a lot of product is lost in purification
http://www.sciencemadness.org/talk/viewthread.php?tid=19378#...
| Quote: | | Also HN3 makes me a bit nervous, my fumehood fan isn't what it used to be. |
but it would form in-situ and get
used up immediately.
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
OK, I think I’m going to pursue this one. I have little access to my fumehood at the moment (I have to travel for an hour), and this one looks like
I could do it at home as there is no heating, distillation and there aren’t very corrosive chemicals, as I want to do the Hofmann rearrangement with
hypochlorite and HCl is doable with good ventilation.
Also I already have half of the chemicals and I can get the rest.
As a warned man counts for two, I would like to share my plan with you before starting.
My plan is to mostly follow the article from my first post as I the discussion before gave me more confidence in the procedure.
I have a Buchner vacuum filter setup, a magnetic stirrer with stir bars and a couple of beakers and flasks and a separation funnel. Also I can do
rough melting point measurements. A high yield is not my aim so I could do an extra crystallization here or there when I don’t feel sure.
1. Cool a mixture of p-chlorobenzaldehyde and ethyl acetoacetate (1:2.1 molar) on ice and add catalytic piperidine. Stir for an hour and stir for 24
hours at RT. Dilute with 3 volumes of 99+% ethanol. Freeze overnight to crystalize and filter. Wash with 50 % ethanol. Dry in air.
2. Get a melting point, should be around 153oC.
3. A stirred hot solution of 50% potassium hydroxide (density 1.5) is added portionwise to p-chlorobenzylidene-bis- acetoacetic ester (0.15 eq. w/w).
The reaction mixture is stirred and maintained at 90-95 °C for 2 hours (closed flask in water bath), diluted with two volumes of water, washed with
DCM (ether is suggested, but I have DCM), acidified slowly with 0.5 eq. volume of concentrated hydrochloric acid, chilled thoroughly and filtered. The
filter cake is washed several times with ice cold water and dried in an plastic box with dry potassium carbonate as a drying agent (the article
suggests drying in vacuo at 60 °C, but I guess the K2CO3 in a box would suffice).
4. Get a melting point, it should be around 165oC.
5. The next step would be forming the glutarimide, which I want to do as a melt with urea in a microwave. I will try to find a bit more on the molar
ratios and how to purify (probably crystallization) the product. If this doesn’t work I could try the concentrated ammonia, which I have.
6. 125oC should be about it.
7. The glutarimide is mixed 1:1 w/w with NaOH in 6 eq. w/v 12.5%
sodium hypochlorite (cooled) and stirred for 8 hours at RT. A calculated
amount of HCl x0.8 and a bit more is added until pH = 7. Crystals are
recrystallized.
8. Mp 205 oC to go for.
[Edited on 6-2-2019 by Tsjerk]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I have the feeling the first step is going fine. I started as was described in the publication I cited in the first post. I started with a bit more
product than I would normally go for but I had trust in the fact at least the first reaction should work, if so that would give me some material to
work with.
I started the reaction by mixing ethyl acetoacetate (102.1 grams, 100 ml) with 4-chlorobenzaldehyde (53.8 grams, 2.05 eq.) and 5 ml diethylamine. I
cooled everything on ice before adding the catalyst, but I don't think this is necessary because I didn't notice any exothermic reaction and the
reaction is slow as a brick anyway. I ran the reaction for 24 hours, diluted with 400 ml 99% ethanol and cooled to -20 for 12 hours. A bit of white
solids precipitated on the bottom of the beaker. I was a bit afraid this could be water, but it stayed solid after getting above 0, so apparently not.
Now I was getting a bit worried whether this could be 4-chlorobenzoic acid as I'm not running this in a closed vessel. I don't think so because this
compound is highly soluble in ethanol it appears. My concerns were taken away for the most part when I saw the precipitate starting to build up. I'm
only getting photo's of it every 24 hours because I'm not physically there (my mom is getting the photo's  , but the photo's are promising. , but the photo's are promising.

After 24 hour run, diluting with ethanol, 12 hour freeze at -20 and getting back to ambient temperature (8-10 degrees).

After running for another 72 hours.

after 120 hours
[Edited on 12-3-2019 by Tsjerk]
[Edited on 13-3-2019 by Tsjerk]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Nothing seems to be happening anymore. I will filter and wash with 50% ethanol tomorrow evening. Let's see what we've got!
Depending on the speed of drying I can maybe already do the decarboxylation with NaOH tomorrow... I think I will suspend the material in water and add
the NaOH prills to that suspension while stirring. That will probably heat up a bit but I guess that is more controllable than adding a concentrated
NaOH (actually KOH is suggested) solution to the solid material which is suggested in the paper.
[Edited on 13-3-2019 by Tsjerk]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I think the deacetylation and hydrolyses worked. The yield of the first step was only 22%, but that still gave me 33 grams to work with.
I dissolved 79 grams of NaOH in 225 ml of water and poured that over the compound of reaction one. The mixture turned bright yellow, that color
disappeared after about an hour and after one and a half hours a drop of the reaction mixture cleanly dissolved in cold water. Most solids dissolved
after adding two volumes of water to give a dark yellow solution. This was washed with DCM and 0.66 eq volume of 30% HCl was added. The yellow
disappeared and a white solid precipitated. This was filtered and washed with ice water.
Edit: the reaction was done at 90 degrees.
[Edited on 24-3-2019 by Tsjerk]
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
I got to baclofen! 3% overall yield but I already got the first step up from 22% to over 80%! It is still drying but I'm pretty sure it is way over
80%
Details following soon
[Edited on 20-4-2019 by Tsjerk]
|
|
|
DrScrabs
Hazard to Others
Posts: 123
Registered: 13-3-2018
Location: Laputa
Member Is Offline
Mood: Still evaporating..
|
|
Nice! Really excited for the thread!
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
Update on step one: I got a 66% yield during the second try, but during the third try I used 2,5 molair eq ethyl acetoacetate instead of 2.1. The
yellow color is a lot less pronounced and I'm expecting a yield higher than 66%. I can perform the second and the third steps close to quantitative,
at least 90% each.
I'm still struggling with the fourth step. The first time it worked but a lot of HOCl was given of indicating, I think, a lot of the diacid from the
second step still left in the precursor. The reaction also became neutral instead of basic. The first try of the third step didn't went too well,
therefore probably the amount of diacid still present. The second try of the fourth step gave, I think because there was much more precursor, some
intermediate, next time I will use a lot more bleach.
|
|
|
Tsjerk
International Hazard
Posts: 3035
Registered: 20-4-2005
Location: Netherlands
Member Is Offline
Mood: Mood
|
|
p- chlorobenzylidene-bis-acetoacetic ester (compound 2)
57 g 4-chlorobenzaldehyde (0.405 mol) was dissolved in 131 ml of ethyl acetoacetate (1.014 mol, 131,9 g, 2,5 eq.) and 10 ml of di-ethylamine was
added. The mixture was stirred and left to stand at RT for one week. The resulting mass was pulverized and dissolved in 900 ml 50% ethanol/water by
heating up to boiling temperature. After crystalizing at RT the mixture was filtered and washed with 50% ethanol until the filtrate was colorless. To
dry, the filter cake was heated in a big beaker in a microwave at 300 watt for 30 minutes. Yield: 93%; mp: 153-155 degrees.
β(p- chlorophenyl) glutaric acid (compound 3)
33 g (0.086 mol) of compound 2 was added to 80 g NaOH dissolved in 225 ml of water (final NaOH concentration 6.5 M). The suspension was heated on a
100 degrees water bath while insulated to ensure proper heating of the whole mixture. The mixture was not stirred. When carbon dioxide evolution
ceased after one and a half hour, the temperature of the mixture rose from 95 degrees to 100 degrees. 300 ml water was added and the mixture was
heated until most solids dissolved and filtered hot. To the filtrate 50% (v/v) sulfuric acid was added until pH 1 was reached (approximately 150 ml).
The filtrate was cooled to RT and filtered. The filter cake was washed with cold water until the filtrate was pH 7 and dried for 30 minutes in a
microwave at 300 watt. Yield: 80%; mp: 165-167 degrees.
β(p- chlorophenyl) glutarimid (compound 4)
In a 100 ml beaker 10.0 g (0.0412 mol) of compound 3 was mixed with powered urea (2.60 g, 1.05 eq.) and boiling water was added while heating until
all solids dissolved. The solution was boiled to dryness and the precipitate was heated in a microwave at 850 watt until the melted mass stopped
giving of gaseous reaction products (approximately 6-7 minutes). The still molten mass was dissolved in a minimum of 33% ethanol/water at boiling
temperature. The solution was filtered hot and after crystallizing at RT the filter cake was recrystallized from 33% ethanol/water and the product
dried in air. Yield: 75%; mp: 124-126 degrees.
Amino-3- (4-chlorophenyl) butanoic acid (baclofen)
3.85 g (0.0962 mol) NaOH in 33 ml of water was added to 33 ml 12,5% (0.066 mol) sodium hypochlorite and cooled on ice. 11.0 g (0.0493 mol) of compound
4 was slowly added and the mixture was stirred for 10 minutes on ice and 8 hour at RT. To this 2.10 grams of sodium sulfite (0.0180 mol) in 10 ml
water was added. The mixture was carefully adjusted with 3% w/v HCl to pH 7.5 and cooled on ice. The mixture was filtered and the filter cake washed
with little ice cold water. The cake was recrystallized from water and dried for 30 minutes in a microwave at 300 watt. Yield: 33%; mp:203-205
degrees.
I screwed up the recrystallization of the baclofen, so I think that 33% could be raised significantly.
*acetoacetic ester: The synthesis of compound 1 can be done almost quantitatively with ethyl acetoacetate being used in a 2,5 molar eq. quantity and
a 10% molar amount of di-ethylamine. Probably other secondary amines can be used in a similar fashion. The reaction mixture is crystalized from 6
volumes of 50% ethanol in water and washed with 50% ethanol in water until white.
**Diacid: compound 2 is synthesized by decarboxylation and deacetylation in 6.5 molar NaOH at boiling water bath temperatures. Compound 1 is finely
ground and added to cold 7 molar NaOH and heated to 100 degrees for 2 hours. 2 eq. volumes of boiling water are added and the mixture is heated until
most solids dissolve. The mixture is filtered hot and cooled to RT. 50% v/v sulfuric acid is added until pH 3 and the mixture is cooled again to RT.
After filtering the filter cake is washed with cold water until the filtrate is pH neutral.
***glutarimide: This product was prepared via the di-ammonium salt and the mono-urea salt of compound 3. Both need heating until melting
(approximately 200 degrees) to liberate water/ammonia in order to form the glutarimide. It was found heating in an oven or on a heating plate of the
two salts gave comparable yields at around 30%. Microwave irradiation at 850 watt of the di-ammonium salt gave a yield of 50%. The microwave treatment
of the mono-urea salt, 6 minutes at 850 watt, gave a yield of 77%. In all cases the mixture was heated until no bubbles were given off.
****baclofen: pH 7.5 seems to be very important for a proper yield as the product dissolves at anything above pH 8 or below pH 7.
[Edited on 25-5-2019 by Tsjerk]
|
|
|








