Klute
International Hazard

Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
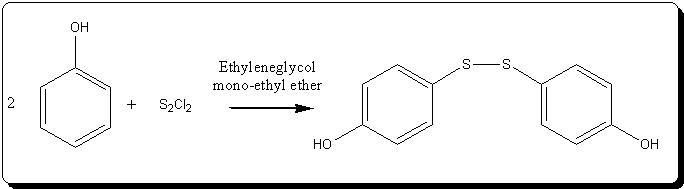
Synthesis of bis(4-hydroxyphenyl)disulfide and alkylated derivatives
Phenols react with sulfur mono-chloride to form symetric hydroxyphenyldisulfides. Without any substituants in para- position, the disulfide
bond forms predominantly in para-.
Several side reactions can accur: formation of ortho- isomer, formation of chlorophenols, and disproportion of the disulfides to symetric
mono-sulfides and elemental sulfur. According to EP0287292, using ethylenglycol mono-alkyl ethers, these side-reactions are substancially reduced. I tried this reaction with Ethyleneglycol
mono-ethyl ether (EEG).
Sulfuration of phenol
In a 250mL 4-neck RBF, equipped with a condenser connected to a 30% NaOH wash bottle, a addition funnel connected to a argon inlet, a thermometer and
magnetic stirring, 28.52g (303.05 mmol) of phenol are introduced, followed by 125mL EEG. The dissolution takes a minute and gives a slightly
salmon-colored limpid solution.




22.5g (166.67 mmol) of freshly distilled S2Cl2 are charged in the addition funnel, and the setup flushed with argon.

The sulfur mono-chloride is then slowly dripped in the phenol solution, at 15°C. The reaction is slightly exothermic. The reaction mixture immediatly
takes a golden orange colour, and darkens as the addition continues. The temperature increases up to 35°C, a cold water bath is applied to keep the
temperature at 25°C. The addition takes 30min, there is a slow evolution of HCl gas. A dark brown/red solution is obtained.
5min after beggining of addition:

20min after:

The flask is then heated at 50°C for 30min. Evolution of HCL increases, small bubbles at seen evolving from the reaction mixture. When the oil bath
is removed, the reaction mixture has cleared up to a golden yellow color.

The flask is cooled to 25°C, and it's contents are drowned in 400mL of dH2O. A canary yellow milky emulsion is obtained, with a very thick, viscous
bright yellow oil at the bottom. 50mL of DCM are added to dissolve the viscous material, and the beaker's contents are transfered to a 1L seperating
funnel. The organics are seperated, and the aq extracted with 3x50mL DCM. The aq clears up entirely after the first extraction, the extract been clear
yellow. The combined organics are washed with 2x100mL dH2O and 100mL 1/2 saturated brine. The extract is then dried over Na2SO4 and placed in the
freezer overnight.



The solution is then filtered through a cotton plug into a 500mL distn flask, the dessicant rinsed with a little fresh DCM. The solvant is then
removed under atm pressure. The orange residu obtained is kept under vacuum for 30min to remove solvant remains. The crude product weighs 61g.
Apparently, there still is some EEG left in the product. After cooling in the fridge, it has thickened even more, as very thick honey. I have no idea
of how i'm going to weigh portions out. Hopefully it will fluidize enough on mild warming.
Distn setup:

Still:

Residu under vacuum:

Crude product:

TLC shows there is a little starting material, but seperation of the spots is very sparse. Several solvent systems have been tried, the best yet been
20% pet ether in AcOEt, showing two spots.
Comments
The addition took a little more time that is indicated in the patent, and the temperature increased a little too much (35°C) by moments. Next time,
the cold procedure might be tried. I guess a few more minutes heating could have helped, as there still was a little HCl evolution.
The absence of sulfur deposit is good sign, the dreaded disproportion seemed to have been avoided.
The disulfide is now stored under argon in the fridge, some will be O-alkylated, some will be S-alkylated, and some will be totally alkylated  I might try formylating some o-alkylthiophenol if i get enough time. I might try formylating some o-alkylthiophenol if i get enough time.
For the O-alkylation, i will be using NaH as a base in DMF and TMP or EtBr as i have these at hand, but considering there must be some EEG left,
should I use a little excess base? It might reduce the disulfide bond if given enough time... But i guess the phenolic -OH are much more acidic than
the EEG's hydroxy, so i might consider ~95% yield and use adequate amount of NaH, counting ~5% excess to completly dry the DMF. I guess the NaH would
react with any EEG before it attacked the disulfide bond.. Any advise welcome!
Further details conserning the alkylations to come.
EDIT: Decided on performing the O-alkylation tomorow, it's too late for tonight. I will be using what's left of my MeI, just enough for 100 mmol of
phenolic compound.
[Edited on 16-4-2008 by Klute]
[Edited on 17-4-2008 by Klute]
|
|
|
garage chemist
chemical wizard
Posts: 1803
Registered: 16-8-2004
Location: Germany
Member Is Offline
Mood: No Mood
|
|
Very interesting what you are doing with S2Cl2.
Will you try to cleave the S-S bond to make p-hydroxythiophenol?
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Actually, I will try to directly alkylate the S by insitu reduction of the disulfide and direct S-alkylation, one pot. This can be done with either Zn
in DMF, followed by RX, or Rongalite/RX/K2CO3 (that's why i looked into the preparation of this compound). There are several references available in
the ref forum.
I will try to isolate some O-alkylthiophenol though. Not too sure how bad it's going to smell. If it's in the same range as S2Cl2, it's bearable.
I'm thinking of formylating it with Mg(OMe)2/Paraformaldehyde, but that's just an idea form now. I'm going for asymetric alkoxyphenylthioethers right
now (O-alkylation of the hydroxyphenyldisulfide, followed by one-pot reduction/alkylation of the sulfur).
I'm impressed at how well I'm getting out at doing lab work with one hand I'm
been very carefull though, I must admit I'm breaking more glassware that i usually do 
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
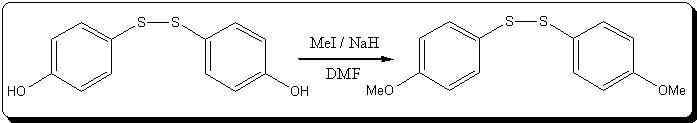
16.5g of the previous crude viscous disulfide is weighed into a 250mL schlenk (containing 40mmol disulfide if considering 100% yield in the previous
reaction), and 70mL DMF are added. The oil takes a few minutes of stirring to completly dissolve. The schlenk is purged two times, and maintained
under argon.

3.23g (80 mmol) of 60% NaH dispersion in mineral oil are weighed out and placed in a oven-dried 250mL 4-neck RBF. A addition funnel and condenser,
respectively connected to a argon and vacuum inlet, are attached.




30mL of DMF are added to the hydride, causing some bubbling (6 months old DMF). The suspension is breifly purged two times, and a bubbler is
attached on top of the condenser in place of the vacuum inlet.

The disulfide solution is transfered to the addition funnel via canula, and the flask cooled to 10°C with a ice water bath.

The disulfide is then slowly dripped in the hydride suspension with steady stirring. H2 evolution starts immediatly, and the grey suspension turns
brown, gradually darkening as the addition continues. The drip rate is regulated to keep the temperature under 13°C and keep a constant H2 evolution
(1 drop/sec). The schlenk and the addition funnel are rinsed with 10mL DMF.




Once the addition is complete, stirring at 10°C is continued until no more H2 evolves (20min).
5mL (80.32 mmol) of MeI are then introduced in the addition funnel via syringe with adequate precautions. The alkyl iodide is then dripped in over 5
min, the container and addition funnel are rinsed with 10mL DMF.
An exothermic reaction sets in, with deposition of red solids, to a point where stirring is difficult. The thick slurry is gradually broken up to a
more fluid suspension by magnetic stirring. After having heated up to 25°C, the temperature falls back to 10°C. The cold water bath is then removed,
and the flask left to stir for 45min.

The flask contents are then drowned in 450mL brine, giving a coffee-colored suspension, with a very viscous brown semi solid paste floating on the
surface. After 5 min with occasional stirring, the mixture is transfered to a seperating funnel, with 100mL DCM. The viscous material is dissolved,
and the dark red/black organic layer seperated. The orange aqueous layer is extracted with 75+2x50mL DCM, giving a emulsion each time taking a few
minutes to seperate.


The combined organics are then washed with 150mL 3% NaOH solution. A opaque milky yellow aq seperates slowly out, clearing the organic layer a bit.
The organics are then washed with 3x100mL dH2O, the washes comming out yellow, and 100mL 1/2 saturated brine (slightly yellow). The dark red solution
is then dried over Na2So4.

The basic extract and the water washes are combined then acidified with conc HCl. It forms a yellowish milky solution, with brown oily droplets. The
initial DMF/brine aq layer is also acidified, giving a pink milky solution, with a little brown/red oil at the bottom. The two aq are extracted with
3x15mL DCM each, giving a ruby red solution, that is washed with 2x50mL h2O (clear), and 100mL 1/2 sat brine (clear). The acidic extract is then dried
over Na2SO4.
Basic extract/washes after acidification:

DMF/Brine after acidification:

Acidic extract:

TLC (5% AcOEt in pet ether) indicates the neutral extract still contains the products present in the acidic extract (3 very close spots), and a
larger spot surely the methylated disulfide. The acidic extract could contain: unreacted hydroxyphenyldisulfide, hydroxthiophenol, and it's mono S- or
O- methylated derivatives, aswell as impurities from the sulfuration reaction.
Coluum chromatography could be very helpfull here, as the disulfide spot in largely above the impurites, grouped up together.
The neutral extract was transfered to a distillation setup, and DCM is currently being removed. Evaporation of the solvant in the funnel left a solid
crust, so the product seems to be a solid at RT. Hopefully a recrystallization or two will remove most of the impurities.
The acidic extract will also be evaporated, and if there is enough product, possibly methylated with TMP to try and recover p-methoxyphenyl methyl
sulfide. I guess the major side reaction would be reduction of the disulfide.
EDIT: a dark brown oil was obtained, covered with a little layer of clear liquid, that would go even with mild vacuum applied for some time at
50°C. The oil was cooled, some the scrapped powder from the funnel added, no cristallization. 15mL of pet ether were added, and the flask put in the
freezer overnight, still no cristallization... I might consider purifying a sample with a coluum, but having only home-made silica at hand (and not
much), i will never be able to purify all of it.. I'm surprised that a s olid was left in the funnel upon evaporation though. I might try to dissolve
the oil and wash it with some more dilute NaOH to try and remove as much impurities as possible...
Any advise welcome. I could use the oil as is for reduction of the disulfide bond, but that would be 2 reaction in a row using crude product, it's
asking for trouble... and i'm not even sure if the end product is a solid or not.
Could anyone with acces to a more elaborate chemical database than .. google help me out here? I've got a few similar compouds I'd like to know the
mp's and bp's, that i just can't find screening through random web pages...
Related discussion: http://www.sciencemadness.org/talk/viewthread.php?tid=10291
[Edited on 18-4-2008 by Klute]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
UPDATE: a stupid attempt at distilling the crude brown oil gave a fraction boiling from 70 to 130°C at 25torr, which turns out to be surely anisol,
from unreacted phenol which got methylated. Next time i perform a sulfuration, i will leave it a little longer, and use a very slight excess S2Cl2.
After that, by 200-230°C, a dark orange/red vicous oil started passing, but only 8mL were collected before the residu turned to solid tar: crunchy
coal-like carbon mass... And the distillate is contaminated by anisol (TLC: 20% DCM in pet ether, prefer 5% AcOEt in pet ether). Then i finally found
a bp for the compound: 230°C at 0.4torr.... I was not even close to collecting the pur product...
So this is left down for the moment.
Reduction of bis(4-hydroxyphenyl)disulfide to 4-hydroxythiophenol
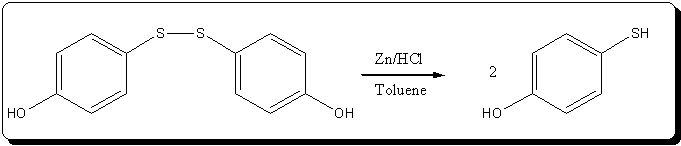
27g of crude bis(4-hydroxyphenyl)disulfide oil (containing at most 130 mmol) were transfered to a 250mL 4-neck RBF with a stir bar. 75mL of toluene
were added, it took some heating to fluidize the oil enough for the stirring to start, and after 5 min strong stirring, a homogeneous orange solution
was obtained.



8.25g (126 mmol) of zinc dust were weighed, and added to the stirred solution of the disulfide at room temp. The toluene solution took a green tint
immediatly. The suspension was heated up to 50°C in a oil bath with strong stirring.


37.5 mL (355 mmol) of 30% HCl were then slowly dripped in, causing vigorous H2 evolution, and a exothermic reaction: the temp increased to 65°C. The
green color quickly turned yellow, and the zinc dust stayed in the bubbles of the acid, with strong foamming. The drip rate was ajusted to have a
controlled reaction, without excessive foamming or gas evolution ~1drop/2sec) . After 20min, the oil bath was removed, and a few minutes after the H2
evolution sudenly ended. The rest of the acid (~5 mL) was added in quickly, and the oil bath replaced. Heating and stirring were continued for 1H.



UPDATE:
Pfffff....... A few seconds after transfering the flask's contents to a seperating funnel, copious amouts of viscous toluene-insoluble yellow
gel/paste started depositing inside.. After a first water wash, it was clear than all the thiophenol had been oxidized to the disulfide (yellow gak,
soluble in acetone), by the disappearance of the strong rotten-eggs smell, and clearing up of the yellow color in the toluene layer... I really don't
understand how the patent could have done 3 extractions without this happening! A workup under inert atmospher is not concivable.
The disulfide was dissolved in acetone, at least i seperated it from the other impurites... but i don't understand how i manadged to dissolve it in
75mL toluene in the first place?! pfff...
I think i'm going back the TEMPO project for the few days i have left.. less smelly, less mess, and less failure ! I will try a different approche on the sulfides, and never try to isolate a
thiophenol again... Insitu reaction or nothing!
[Edited on 20-4-2008 by Klute]
|
|
|
|








