| Pages:
1
..
74
75
76
77
78
..
104 |
sparkgap
International Hazard

Posts: 1234
Registered: 16-1-2005
Location: not where you think
Member Is Offline
Mood: chaotropic
|
|
Quote: Originally posted by gluon47  | My school has given me the opportunity to do a few experiments in there lab. I noticed they had a bottle of anhydrous propionic acid, so I thought I
might make a propionate ester.
I would like to try isopropyl propionate. Fischer esterification should work for making this ester right?
Would adding anhydrous magnesium sulphate to the reaction mixture during reflux help to increase yield?
Any advice would be much appreciated. |
Fischer should work for both primary and secondary alcohols. Isopropyl alcohol is cheaper than propionic acid, so use that in excess. If you can use a
Dean-Stark trap, do so; magnesium sulfate won't do much in those reaction conditions.
sparky (~_~)
"What's UTFSE? I keep hearing about it, but I can't be arsed to search for the answer..."
|
|
|
mesanaw
Harmless
Posts: 10
Registered: 29-10-2016
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by gluon47 |
I would like to try isopropyl propionate. Fischer esterification should work for making this ester right?
Would adding anhydrous magnesium sulphate to the reaction mixture during reflux help to increase yield?
|
Yes, a Fischer esterification would occur. Considering you are already using anhydrous propionic acid and presumably 99% isopropanol, the use of
magnesium sulfate during reflux would not materially affect yield. I have seen desiccants used after reflux and separation in order to remove the
residual water, but not during reflux.
[Edited on 19-6-2017 by mesanaw]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
I don't think I'd put any desiccant in the reaction mixture itself, but you might want to put some in the reflux path: https://www.youtube.com/watch?v=Ah5ds_3s5BI
Magnesium sulfate might clump up if you put it in a Soxhlet and run wet solvents through it, so molecular sieves are probably better.
|
|
|
gluon47
Hazard to Self
Posts: 81
Registered: 20-9-2015
Location: oceania
Member Is Offline
Mood: fluorinated and dying
|
|
Awesome, thanks! . I wont use MgSO4 . I wont use MgSO4
reality is an illusion 
|
|
|
gluon47
Hazard to Self
Posts: 81
Registered: 20-9-2015
Location: oceania
Member Is Offline
Mood: fluorinated and dying
|
|
Performed the reaction today on a 1/6 mole scale with 20% excess of isopropanol. It was interesting to smell the propionic acid. A lot like glacial
acetic acid, but more rancid, I like it.
Isopropyl propionate seems to have the characteristic sweet odour of most simple esters, but quite reminiscent of glue and slightly less sweet.
I'm going back to the lab next week for workup and purification. Can't wait!
reality is an illusion
|
|
|
Geocachmaster
Hazard to Others
Posts: 146
Registered: 5-3-2016
Location: Maine, USA
Member Is Offline
Mood: Corroded, just like my spatulas
|
|
Saccharin
The first photo is me trying to make sense of the pathway from methyl anthranilate to saccharin found on Wikipedia, shown in picture two. I don't have access to the reference given or any other papers I find about the synthesis of saccharin.
The diazotization in step one seems pretty straightforward. In step two the diazonium compound is reacted with sulfur dioxide to produce the sulfonyl
chloride at number three. The Wikipedia page on sulfonyl halides says that phenyldiazonium chloride reacts with sulfur dioxide and HCl, so I'm assuming setp two needs an acid
catalyst. In step three the sulfonyl halide is reduced by SO2 to the sulfinic acid. Steps two and three would be carried out together and
at low temperatures because the sulfonyl chloride will react with water. Extra SO2 will exclude air and prevent oxidation of the sulfinic
acid. What I'm confused most about is the two next steps. Wikipedia just says Cl2 and NH3. I read that sulfonamides can be prepared by reaction of an NHR2 with a sulfonyl chloride. For this reason I assumed that chlorine would
react at step four to remake the sulfonyl chloride seen at step three. This would then react with ammonia to produce the sulfonamide. After this I'm
thiking that the H+ on wiki is a hydrolysis which is followed by a ring forming step which must happen automatically.
Does anyone with more experiance than me think this is plausible/makes sense? I think making an artificial sweetener that is 300x sweeter than sugar
would be really cool! It's something I want to do in the future. I'll be purchasing 250g (or maybe 1000g!) of phthalic anhydride and that would be my
starting point.
Any input is greatly appreciated
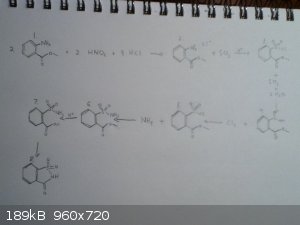 
Sorry for the bad quality  , you have to open the pic to actually see anything... , you have to open the pic to actually see anything...
|
|
|
UC235
National Hazard
Posts: 565
Registered: 28-12-2014
Member Is Offline
Mood: No Mood
|
|
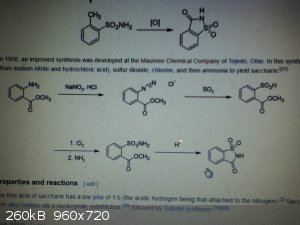
| Quote: Originally posted by Geocachmaster | The first photo is me trying to make sense of the pathway from methyl anthranilate to saccharin found on Wikipedia, shown in picture two. I don't have access to the reference given or any other papers I find about the synthesis of saccharin.
The diazotization in step one seems pretty straightforward. In step two the diazonium compound is reacted with sulfur dioxide to produce the sulfonyl
chloride at number three. The Wikipedia page on sulfonyl halides says that phenyldiazonium chloride reacts with sulfur dioxide and HCl, so I'm assuming setp two needs an acid
catalyst. In step three the sulfonyl halide is reduced by SO2 to the sulfinic acid. Steps two and three would be carried out together and
at low temperatures because the sulfonyl chloride will react with water. Extra SO2 will exclude air and prevent oxidation of the sulfinic
acid. What I'm confused most about is the two next steps. Wikipedia just says Cl2 and NH3. I read that sulfonamides can be prepared by reaction of an NHR2 with a sulfonyl chloride. For this reason I assumed that chlorine would
react at step four to remake the sulfonyl chloride seen at step three. This would then react with ammonia to produce the sulfonamide. After this I'm
thiking that the H+ on wiki is a hydrolysis which is followed by a ring forming step which must happen automatically.
Does anyone with more experiance than me think this is plausible/makes sense? I think making an artificial sweetener that is 300x sweeter than sugar
would be really cool! It's something I want to do in the future. I'll be purchasing 250g (or maybe 1000g!) of phthalic anhydride and that would be my
starting point.
Any input is greatly appreciated
Sorry for the bad quality , you have to open the pic to actually see anything...
|
Sulfinates can be chlorinated to sulfonyl chlorides. A similar reaction occurs during chlorination of bunte salts (see: http://www.sciencemadness.org/talk/viewthread.php?tid=9921&a...). Hydrolysis of the intermediate sulfur chlorides ends with a modestly stable
sulfonyl chloride.
Treatment with ammonia gives the sulfonamide.
If you're only after a few grams, saccharin is readily isolated from US Sweet-n-low (or equivalent off-brands) where it is present as its sodium salt
at roughly 3.5% by weight. The solid sweetner is dissolved in roughly twice it's weight in water, filtered to remove anticaking agents, and acidified
with HCl. The saccharin free acid precipitates as a white powder (I recommend chilling it in an ice bath or fridge for a while). Vacuum filter off the
liquid and rinse with ice water to remove contaminating glucose.
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
Does anyone know offhand where to get a few grams of n-phenylanthranilic acid?
|
|
|
TheNerdyFarmer
Hazard to Others
Posts: 131
Registered: 30-9-2016
Member Is Offline
Mood: No Mood
|
|
Question
Does anyone know of a relatively cheap source of absolute ethanol? I am having trouble finding good affordable sources of it. It would also be nice if
there was an affordable source of lower concentration that I could dry myself.
|
|
|
Morgan
International Hazard
Posts: 1694
Registered: 28-12-2010
Member Is Offline
Mood: No Mood
|
|
This was $12.99 for 750ml, a couple of dollars cheaper than the other 190 proof Everclear next to it in the store.
http://wongdrinks.blogspot.com/2012/04/i-drank-that-clear-sp...
|
|
|
j_sum1
Administrator
Posts: 6320
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Oxygen-containing cations
Just a curiosity mostly.
Oxyanions abound. But are there any polyatomic cations that contain the element oxygen?
I could not think of any but then I have not had much dealing with polyatomic cations except for ammonium.
|
|
|
ninhydric1
Hazard to Others
Posts: 345
Registered: 21-4-2017
Location: Western US
Member Is Offline
Mood: Bleached
|
|
j_sum1 do complexes count? Because [Co(H2O)6]2+ is a 'cation' that contains water, which in turn contains oxygen.
True polyatomic cations containing oxygen are probably impossible due oxygen's high electronegativity.
EDIT: Never mind, Harristotle has some.
[Edited on 7-13-2017 by ninhydric1]
[Edited on 7-13-2017 by ninhydric1]
|
|
|
Harristotle
Hazard to Others
Posts: 138
Registered: 30-10-2011
Location: Tinkerville
Member Is Offline
Mood: I tink therefore I am
|
|
Vanadyl and Uranyl are the ones that spring to mind VO2+ and UO22+.
Other than that, dunno.
|
|
|
j_sum1
Administrator
Posts: 6320
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Strange purple chromium compound
I am always cautious with hexavalent chromium and like to reduce it to Cr(III) before adding to the Cr waste bucket. Today I was cleaning up some
solutions and observed a transient purple colour that I have not seen before. Details follow:
This was a demonstration I did for my students on oxidation states. We began with solutions of trivalent and hexavalent chromium, oxidised one and
reduced the other to have their colours switch.
The solution in question began as chromium sulfate, Cr2(SO4)3 -- about 10mg dissolved in 40mL of water.
A few pearls of sodium hydroxide were added and dissolved to make the solution alkaline. Nothing was measured but the NaOH was well in excess
of what was required.
Then a splash of hydrogen peroxide was added and over a short period of time the orange colour of hexavalent was observed. It looked more like
dichromate than chromate -- which on reflection seems odd since the solution was very alkaline and not particularly concentrated.
After sitting for a couple of hours I began to clean up this and other experiments -- using mostly what was on hand. I added a few crystals of
sodium thiosulfate and swirled them around but they were not completely dissolved. No change occurred since the solution was too alkaline.
I then added some concentrated sulfuric acid dropwise from a pipette. As the drops fell through the solution they formed a green plume that
dissipated back to orange. At the bottom of the beaker where the thiosulfate concentration was higher a mist of sulfur precipitate was formed.
I then squirted an excess of sulfuric acid in and began to stir. The solution first went cloudy white and then a royal purple colour appeared
for about a second before the mixture turned almost colourless.
I had one of those "what was in there?" moments as I hadn't seen that purple before and was certainly not expecting it.
On reflection I guess it was either a peroxide complex or a transient Cr(IV) compound. Can anyone enlighten me?
|
|
|
j_sum1
Administrator
Posts: 6320
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
Cool. Thanks. I should have thought of those. But since polyatomic cations are a rarity for me my mind just did not gravitate in that direction.
|
|
|
Tdep
National Hazard
Posts: 519
Registered: 31-1-2013
Location: Laser broken since Feb 2020 lol
Member Is Offline
Mood: PhD is done! It isn't good but it's over lol
|
|
| Quote: Originally posted by j_sum1 |
On reflection I guess it was either a peroxide complex or a transient Cr(IV) compound. Can anyone enlighten me? |
Hello!
Blue/purple chromium in the presence of acidified peroxide, that sounds like Chromium(VI) oxide peroxide! I have a gif of me making some on twitter, hopefully you can see it... https://twitter.com/Explosions_Fire/status/87898935016739226...
I'll hopefully have a video up in the next two weeks where I give better footage of it. It's quite unstable in water so only appears transiently, so
it seems to match what you're describing pretty well
|
|
|
j_sum1
Administrator
Posts: 6320
Registered: 4-10-2014
Location: At home
Member Is Offline
Mood: Most of the ducks are in a row
|
|
That looks very much like it. Mine was a lot lighter (concentration) and a bit more towards the purple and was cloudy with the precipitate at the
time.
But it is cool learning new stuff like this. Thanks for that link.
edit: spelling.
[Edited on 13-7-2017 by j_sum1]
|
|
|
JJay
International Hazard
Posts: 3440
Registered: 15-10-2015
Member Is Offline
|
|
It can actually be extracted with ether and used as a high-yielding chromium oxidizer in anhydrous conditions (so it can turn primary alcohols to
aldehydes without proceeding to a carboxylic acid, for example). It forms a somewhat stable adduct with pyridine and a highly stable adduct with bipy.
The bipy adduct can be stored on the shelf for months.
|
|
|
ficolas
Hazard to Others
Posts: 146
Registered: 14-5-2016
Member Is Offline
Mood: No Mood
|
|
In the combustión of an inorgánicos ester, like triethyl borate, what product does the acid part form? The acid again? So, boric acid in the case of
thriethyl borate?
|
|
|
DraconicAcid
International Hazard
Posts: 4332
Registered: 1-2-2013
Location: The tiniest college campus ever....
Member Is Offline
Mood: Semi-victorious.
|
|
| Quote: Originally posted by ficolas | | In the combustión of an inorgánicos ester, like triethyl borate, what product does the acid part form? The acid again? So, boric acid in the case of
thriethyl borate? |
Or just the oxide.
Please remember: "Filtrate" is not a verb.
Write up your lab reports the way your instructor wants them, not the way your ex-instructor wants them.
|
|
|
ficolas
Hazard to Others
Posts: 146
Registered: 14-5-2016
Member Is Offline
Mood: No Mood
|
|
So it would be both, depending on the acid decomposition temperature or something?
And if its the oxide that forms, I guess it would be the oxide the acid decomposes to, right?
|
|
|
ninhydric1
Hazard to Others
Posts: 345
Registered: 21-4-2017
Location: Western US
Member Is Offline
Mood: Bleached
|
|
I plan to get one of these:
http://www.ebay.com/itm/192238825824
in the near future with the tight budget I have.
I'm planning to somehow adapt it to my current non-magnetic stirrer hotplate, so I was wondering how far above the magnetic stirrer would the magnetic
stir bar still spin adequately?
|
|
|
TheNerdyFarmer
Hazard to Others
Posts: 131
Registered: 30-9-2016
Member Is Offline
Mood: No Mood
|
|
I know we have science madness patches for sale on this forum, but do we have things like bumper stickers for sale??
|
|
|
TheNerdyFarmer
Hazard to Others
Posts: 131
Registered: 30-9-2016
Member Is Offline
Mood: No Mood
|
|
Okay, this is something that has confused me for some time. I hear from some people that the Mica window on a Geiger Muller tube enables it to be able
to detect alpha radiation. But now I go on eBay and see some seemingly high quality pancake probes but then I will find out that they can only detect
beta and GT gamma radiation. Does or doesn't the Mica window enable the probe to detect alpha particles??
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
quenching a grignard/NH4Cl substitute
I need to quench a grignard reaction, and in this case the reaction is usually quenched using a saturated ammonium chloride solution.
However, I used all up and need to quench the reaction anyway.
What could be a possible good substitute here? I thought about using ice and diluted HCl, like 5%, but I am not sure, the substrate could be sensitive
to it(an indolylketone, by the way).
|
|
|
| Pages:
1
..
74
75
76
77
78
..
104 |










