| Pages:
1
..
5
6
7 |
chemgirl
Harmless

Posts: 4
Registered: 22-1-2017
Member Is Offline
Mood: No Mood
|
|
So I attempted a "poor person's sublimation" out of curiosity. This works very well for caffeine with a much higher mp/bp, so I figured I'd give it a
go:
0.05g crude tryptamine was placed in the bottom of a glass petri dish and the lid placed on top. On the top of that lid was placed a beaker full of
ice water. All of this was put on a hot plate on low. After a few minutes, the inside of the dish filled with white smoke, and the heat was turned
off and the appartus allowed to cool.
0.0035g of white, spectacularly formed crystals were collected from the underside of the lid, and which smelled like a richer, more aromatic version
of the crude extract. I figured the yield could be explained by the small amount of starting material (static is annoying!) plus oxidation (a certain
amount of charring occurs, which could be limited by doing this in an inerter atmosphere). Regardless, this looked like a great way to make a very
pure analytical standard, and I was so happy to share it with you...
Until I took the MP, which is less than 100c! I will further investigate, but unless my thermometer is just screwed up, I am very confused.
Thanks for the advice, I think I will try this again with more tryptophan and less carvone. And fantastic find. Have you tried it?
[Edited on 29-1-2017 by chemgirl]


[Edited on 29-1-2017 by chemgirl]
|
|
|
alking
Hazard to Others
Posts: 252
Registered: 11-3-2016
Member Is Offline
Mood: No Mood
|
|
Very nice. For an airlock I used an inverted funnel in a beaker of water connected to some tubing. I ran this to a small 2 necked flask connected to
tubing on both sides and this to the top of my reflux condesor. The flask in the middle is to catch any back flow in the case of a pressure drop. If
you do not use this the water will flow back into the reaction flask after the CO2 creation stops and as soon as that water hits the ~200C mineral oil
it will immediately boil causing an explosion.
|
|
|
Mush
National Hazard
Posts: 633
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
POLSKA RZECZPOSPOLITA LUDOWA
URZAD PATENTOWY P.R.L
OPIS PATENTOWY 130769
Zgloszono 80 11 15/ p. 227 894/
int. Cl.3 C07D 209/16
Zgloszonie ogloszono 82 05 24
Opis patentowy opublikowano 1985 12 30
Tworcy wynalazku: marian Eckstein,Stanislaw Misztal,anna Tarczynska,mieczyslaw Adamus.
Uprawniony z patentu: Krakowskie zaklady Farmaceutycne "POLFA", Krakow POLSKA
sposob wytwarzania chlorowodorku Tryptaminy
Przedmiotem wynalazku jest sposob wytwarzania chlorowodorku tryptaminy.
Zany jest z literatury seiichi takano i wspol-pracownikow heterocycles vol. 6,no 8 1977 str. 1167 sposob wytwarzania tryptaminy na drodze
dekarboksylacji tryptofanu w temperaturze wrzenia tetraliny 206°C przy stosunku tryptofanu do tetraliny 1:10 w obecnosci ketonow alifatycznych lub
aldehydow.
Reakcje prowadzono w skali 10-13 g. przy czasie reakcji wynosazym 2-12 godzin uzyskiwano wydajnosci 50-86%
natomiast w skali 0,5 mola zwieksze sie czas ogrzewania 2-2,5 krotnie i podana w przepisie wydajnosc jast nie do odtworzenia.
znana jest takze mozliwosc prowadzenia dekarboksylacji alpha-aminokwasow z publikacji tadashi Suyamai wspolpracownikow C.A 63/7097, 1965 w obecnosci
tetraliny i cyclohexanolu.
W przykladzie podano tylko sposob dekarboksylacji L-izoleucyny w tetralinie z dodatkiem malych ilosci nadtlenku tetraliny.
prowadzenie reakcji dekarboksylacji DL-tryptofanu wedlug znanych sposobow jest uciazliwe z uwagi na prowadzenie reakcji w temperaturze wrzenia
tetraliny ~206°C przy czym uzyskuje sie niskie wydajnosci, zwlaszcza przy powiekszeniu skali procesu.
Nadrodze dlugotrwalych prob opracowano sposob,ktory umozliwia prowadzenie reakcji w temperaturze znacznie ponizej 200°C z wydajnoscia 85-88% w
wsrunkach umozliwiajacych prowadzenie tago procesu rowniez w skali przemyslowej.
sposobem wedlug wynalazku dekarboksylacji D,L-tryptofanu prowadzi sie w srodowisku cycloheksanolu w obacnosci tetraliny zawierajecej nedtlanki
tetraliny lub w obecnosci alpha-tetralonu.
Reakcje prowadzi sie w temperaturze wrzenia mieszaniny,to jest 160-180°C.
Tryptofan zawiesza sie w 2-2,5 krotnej ilosci cycloheksanolu i dodaje tetraline zawierajaca 8-20% nedtlenkow.
Lub alpha-tetralon o zawartosci 80-100%.
Produkt reakcji wydziela sie,po uprzednim rozcienczeniu oziebionej mieszaniny po dekarboksylacji benzenem lub cyklohesanem w stosunku objetosciomym
1:1 w postaci chlorowodorku
przy uzyciu gazowego chlorowodorku lub jego alkoholowego roztworu.
przy procesie dekarboksylacji tryptofanu ilosc nadtlenkow tetraliny w stosunku do tryptofanu winna wynosic 8-12% a ilosc alphatetralonu 6-8%.
PRZYKLAD 1. Dowrzacej zawiessiny 204 g/1 mol/D,L-tryptofanu w 440 g/4,4 mola/ cycloheksanolu wkrapla sie w czasie pol godziny 160 g tetraliny
zawierajacej 9-10% nadtlenku, lub odpowiednio mniejsze ilosc,przy zewarttosci nadtlenkow ponad 10%.
Mieszanine gotuje pod chlodnice zwrotna do uzyskania prawie klarownago rortworu, 3,4-godzin.
Po oziebieniu w atmosferze wydzielajacego sie w reaksji CO2 do 30°C, mieszanine reakcyjna rozciencza sie okolo 440 ml benzenu lub cykloheksanu,seczy
od swentualnej zawiesiny i wytraca chlorowodorek tryptaminy przez dodanie gazowego chlorowodoru lub jego etanolowego roztworu.
Po przemyciu, wydzielonago produktu reakcji,benzenem z dodatkiem 18-20% stanolu i odsaczeniu osadu, uzyskuje sie czysty chlorowodorek tryptaminy o
zawartosci 96-98%,temperaturze topnienia 254-256°C.
Wydajnosc procesu 159-170 g,to jest 82-85% wydajnosci teoretycznej.
PRZYKLAD 2. Do wrzacej zawiesiny 204g/1 mol/D.L-tryptofanu w 440 g/4,4 mola/cykloheksanolu dodaje sie 12 g/0.082 mola/
alpha-tetralonu i prowadzi proces jak w przykladzie 1.
Wydajnosc chlorowodorku tryptaminy o zawartosci 98-99% wynosi 165-173 g, to jest 84-88%.
temperatura topnienia 254-257°C.
Produkty otrrzymane wedlug przykladow 1 i 2 o wzorze sumaryczcym C10H12N2Cl/ciezar czasteczkowy 196.7/sa chromatograficzie jednorodne/w ukladzie
aceton:amoniak, 50 : 0,5/
ZASTRZAZENIE PATENTOWE
1. sposob wytwarzania chlorowodorku tryptaminy z D.L-tryptofanu w srodowisku cykloheksanolu,w obecnosci alpha-tetralonu lub w obecnosci tetraliny
zawierajecej nadtlenki tetraliny,znamienny tym,ze dekarboksylacje prowadzi sie w temperaturze 160-180°C w zawiesinie w 2-2,5 krotnej ilosci
cykloheksanolu w stosunku do D,L-tryptofanu w obecnosci alphatetralonu o zawartosci 8-100% lub w obecnosci tetraliny zawierajacej 8-20% nadtlenkow
tetraliny,a produkt reakcji wydziela sie jako chlorowodorek przy uzyciu gazowego chlorowodoru ,lub jego alkoholowego roztworu po uprzednim
rozcienczeniu oziebionej mieszaniny benzenem lub cyukloheksanem w stosunku objetosciowym 1:1.
2. sposob wedlug zastrz. 1,znamienny tym,ze ilosc dodawanych do reakcji nadtlenkow tetraliny wynosi 8-12% w stsunku do tryptofanu.
3.sposob wedlug zastrz.2 znamienny tym,ze ilosc dodawego do reakcji alphatetralonu wynosi6-8% w stosunku do tryptofanu.
This invention relates to a method for the manufacture of tryptamine hydrochloride.
The method for making tryptamine by decarboxylation of tryptophan at the boiling point of tetralin 206 ° C at a ratio of tryptophan to tetralin 1:10
in the presence of aliphatic ketones or aldehydes.
Reactions were carried out on a scale of 10-13 g with a reaction time of 2-12 hours yielding a yield of 50-86%
On a scale of 0.5 moles the heating time will increase by 2-2.5 times and the stated capacity will not be reproduced.
Also known is the ability to conduct alpha-amino acid decarboxylation from Tadashi Suyamai Associates C.A 63/7097, 1965 in the presence of tetralin
and cyclohexanol.
In this example, only decarboxylation of L-isoleucine in tetralin with small amounts of tetralin peroxide is given.
Running the DL-tryptophan decarboxylation according to known methods is difficult due to the reaction of tetralin at 206 ° C, which results in low
yields, especially when the scale of the process is increased.
Long-lasting experiments have developed a method that allows the reaction to proceed at temperatures well below 200 ° C with an efficiency of 85-88%
in the conductors allowing the process to run on an industrial scale.
The decarboxylation of D, L-tryptophan is carried out in cyclohexanol tetrazine tetrazine tetrahydrate or in the presence of alpha-tetralone according
to the invention.
The reaction is carried out at the boiling point of the mixture, i.e. 160-180 ° C.
Tryptophan is suspended in 2-2.5 times the amount of cyclohexanol and tetraline is added with 8-20% nidoxide.
Or alpha-tetralone with 80-100% content.
The reaction product is isolated after dilution of the chilled mixture after decarboxylation with benzene or cyclohexane in a 1: 1 ratio in the form
of the hydrochloride salt
Using a gaseous hydrochloride or alcoholic solution.
By the process of tryptophan decarboxylation the amount of tetralin peroxide in relation to tryptophan should be 8-12% and the amount of
alphatetralone 6-8%.
EXAMPLE 1. 204 g / 1 mol / L, L-tryptophan in 440 g (4.4 mole) / cyclohexanol is added dropwise over a period of 160 g of tetralin containing 9-10%
peroxide, or, respectively, smaller amounts of peroxide More than 10%.
The mixture cooks under reflux coolers until almost 3.4 ° clear.
After cooling in a CO2 recharge atmosphere to 30 ° C, the reaction mixture is diluted with about 440 ml of benzene or cyclohexane, washed with a
slurry and triturated with tryptamine hydrochloride by addition of gaseous hydrogen chloride or ethanolic solution.
After washing, the separated product of the reaction, benzene with 18-20% stanol and the precipitate is collected, pure tryptamine hydrochloride
having a concentration of 96-98%, melting point 254-256 ° C.
Process efficiency is 159-170 g, that is 82-85% theoretical efficiency.
EXAMPLE 2 To a boiling slurry of 204 g (1 mole) of D.L-tryptophan in 440 g (4.4 mole) of cyclohexanol is added 12 g (0.082 mole)
Alpha-tetralone and performs the process as in Example 1.
The yield of tryptamine hydrochloride with a content of 98-99% is 165-173 g, that is 84-88%.
Melting point 254-257 ° C.
Products kept in accordance with Examples 1 and 2 of C10H12N2Cl, volumetric mass 196.7 / s are homogeneous in acetone: ammonia, 50: 0.5 /
PATENT REMARKS
1. a method of producing tryptamine hydrochloride from DL-tryptophan in a cyclohexanol environment in the presence of alpha-tetralone or in the
presence of tetralin containing tetralin peroxides, characterized in that the decarboxylation is carried out at a temperature of 160-180 ° C in a
suspension of 2-2.5 times The amount of cyclohexanol with respect to D, L-tryptophan in the presence of alphatetralone of 8-100% or in the presence of
tetralin containing 8-20% tetralin peroxide and the reaction product is isolated as hydrochloric acid with hydrogen chloride gas or its alcoholic
solution after dilution The chilled mixture of benzene or cyanohexane in a 1: 1 volume ratio.
2. The method according to claim 1. 1, characterized in that the amount of tetralin peroxide added to the reaction is 8-12% in tryptophan.
3. The method of claim 2, wherein the amount added to the alphatetralone reaction is 6-8% relative to tryptophan.
|
|
|
Mush
National Hazard
Posts: 633
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by 12332123  | I have some success to report...
I refluxed 5g of tryptophan and 22g of naphthalene with 2ml of peppermint oil under a dry condensor with vigorous stirring till the reaction went
clear (~1 hour). On cooling a dark amber glass was formed which was dissolved in around 300ml of DCM. The solution was filtered under gravity and then
cooled in an ice bath. I then gassed it with CO2, giving a fine tan precipitate of Tryptamine carbonate which was filtered off and washed with several
small portions of DCM. This was then warmed in around 200ml of anhydrous isopropanol and fumaric acid was added until no more fizzing was observed.
Tryptamine fumarate was collected and washed with several small portions of isopropanol and DCM, giving a nearly white fine powder. Yield ~70%. 
[Edited on 12-4-2011 by 12332123] |
What you have managed to prepare by gassing is carbamate not carbonate.
|
|
|
joe_chem
Harmless
Posts: 3
Registered: 25-1-2018
Member Is Offline
Mood: No Mood
|
|
TA purification via oxalate
Here are my experiences about tryptophan decarboxylation and purification via oxalate
1) Decarboxylation: 40 g tryptophan (99+) + 240 mL turpentine oil + 8 mL MEK + 40 droplets of carvone. 2 x 12 hours under reflux.
Color was dark-brown-red.
Used AB purification: content after decarboxylation was extracted in 60-80 mL parts by 10% HAc (at 80C), total amount HAc was 400 mL. Juicy extract
was filtered.
100 mL part of HAc extract was extracted by CHCl3 (3 x 15 mL), bottom part was dark brown, almost black, which was poured out.
100 mL purified solution was cooled to 0 C and mixed with ~ 15 mL of 50% NaOH (0 C) to pH about 12. Tryptamine base (TA) precipitated and created
bottom layer, put to freezer (-20C)
and after 1 hour, it started to crystallize. When all TA was solid, content was crumbled by knife, cleaned by cool water, 5% solution of NH4OH to
remove rest of NaOH, filtrated and dried above silikagel.
From 40g of tryptophan was prepared 16.4 g raw tryptamine.
Purity of tryptamine varied. In first case (before), when I worked with 10g of tryptophan (250 ml flask), I obtained purity about 93% with yield
47-53%. In this case above (4000 mL flask), purity was much worse, about 82%. I just guess, I boiled tryptophan too long and burned it. Following
purification was performed with this 82% tryptamine.
2) Purification of tryptamine - recrystallization
First, I tried to recrystallize from acetonitrile. Solution was dark-red-brown, used carcoal but color was still quite dark. Cooled to -20C, used
small TA crystal to start. From 1g TA obtained
just 0.1 purified. That was small yield, but purpose was to obtain TA (to be assured that I have pure TA for next tests). Purity was 97.2 % by GC-MS.
3) Purification of tryptamine via oxalate
Here, some people mentioned oxalate as good method of purification, but nobody brought details. Here follows what I did:
a) 5 g of raw TA (82%) dissolved in 15 mL MeOH. In second beaker, 5 g of oxalic acid, dihydrate was dissolved in 15 mL MeOH.
Mixed. Nothing precipitated, but after freezing to -20C, it started. I let in freezer few hours to be sure that all tryptamine oxalate (TAox) was
solid (and probably some extra oxalic acid too).
Filtrated and flushed by cold MeOH. Obtained about 4g dry TAox, light brownish color.
b) 4g of TAox dissolved in ~130mL of water. At 25C, there were solid parts, so heated to 40-50C, then all was dissolved, tea-like color, no solid
particles observed.
At 50C, TA base was precipitated by 40% NaOH => milk like color. Cooled in freezer (-20C), but not too low, about 0-10C. Obtained nice large
crystals of TA, similar to cotton wool, yellowish color.
Filtrated and carefully flushed by cold water (0 C), dried above silikagel. From 4g, obtained ~ 2g OF PURITY 99.5+% !!!
4) B.p. of TA: raw: 92-95C, ACN recrystallized: 100-102C, oxalate purified: 102-105C. Still pretty far from ideal 114-116C.
I used GC-MS. DB5 column, 30m x 0.25 mm, program 40-300 @ 10C/min, split-less injection, ion trap. Sample dissolved in EtOH.
Here I present 2 pictures:
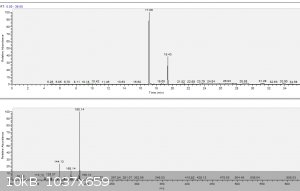
a) TA_raw.png: GC-MS raw TA, purity 82%. 17.09 min is TA, 19.43 min is the only impurity, which I observed. Rest of the peaks are
methyl siloxanes from septum and column. Spectrum of inpurity @19.43 min is on bottom picture. Library suggests following reasonable hits:
1,7-Trimethylene-2,3-dimethylindole
10,10-Spirocyclohexanotetrahydropyrimido[1,2-a]indole
Cycloheptan[a]indole
2-Methyl-1-(4-methylamino-1,3,4,5-tetrahydrobenzo[cd]indol-5-yl)-prop-2-en-1-ol
Carbazole, 1,2,3,4-tetrahydro-3-methyl-
1H-Indole, 4-(3-methyl-2-butenyl)-
...
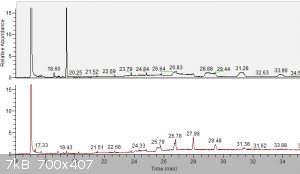
b) TA_purification.png:
Here you can see what changed by oxalate purification. Impurity @19.43min from raw TA (above) was (almost) completely removed.
As before, peaks like 25.79, 26.78, 27.98, 29.48... are cyclic siloxanes (m/z 207, 281 and many other with difference always 74 = Si-O-(CH3)2).
I integrated area for TA (17.09min) and rest of impurity (19.43min) => purity of TA was 99.5%
My conclusions:
1) Even very bad raw TA (82% is really shit) can be converted to excellent purity by simple reaction with cheap
oxallic acid with reasonable yield.
2) There is still space for application of charcoal. My final TA was very lightly brown. Probably in the point 3a) when TA is
dissolved in CH3OH or in 3b) when TAox is dissolved in water.
3) Yield of TA. There is still space for optimizations. In point 3b) where TAox is dissolved in ~130 mL water, it will be probably better to reduce
amount of water and use higher temperature (up to 70C).
Was not successful with purification of TA via carbonate salt. And lazy to buy vacuum pump / apparatus. Hope this will help someone and you will share
experiences with TAox method here.
Enjoy your pure TA 
[Edited on 25-1-2018 by joe_chem]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Interesting. I wonder if the yield of recrystallization might have been better if you had used anhydrous oxalic acid (because water increases the
solubility of most salts). Dehydration kinetics are described in this paper:
http://akademiai.com/doi/abs/10.1007/BF02188864
Unfortunately I do not have access.
|
|
|
Corrosive Joeseph
National Hazard
Posts: 915
Registered: 17-5-2015
Location: The Other Place
Member Is Offline
Mood: Cyclic
|
|
Here ya go.......
'The kinetic study of thermal dehydration of oxalic acid dihydrate'
http://akademiai.com/doi/abs/10.1007/BF02188864
/CJ
Attachment: The kinetic study of thermal dehydration of oxalic acid dihydrate.pdf (380kB)
This file has been downloaded 551 times
Being well adjusted to a sick society is no measure of one's mental health
|
|
|
joe_chem
Harmless
Posts: 3
Registered: 25-1-2018
Member Is Offline
Mood: No Mood
|
|
Thanks guys. Definitely a good point with water in HOx!
In 5g batch of dihydrate, there is 2 g of water and it can affect recovery.
In recipe, I did NOT used saturated solutions, so this is another point what can be improved.
From 5g TA (100%), there should be prepared 7.8 g TAox and from my 82% there still should be obtained 6.4g TAox. So, there is a space to improve.
Here is thread about anhydrous HOx preparation:
http://www.sciencemadness.org/talk/viewthread.php?tid=11153
There are some good points. Seems HOx.2H2O can be slowly dehydrated, some sublimation occurs probably too. I try it just now @95C, look it needs few
hours at least. But for this purpose, some residual water will not be fatal.
[Edited on 26-1-2018 by joe_chem]
|
|
|
joe_chem
Harmless
Posts: 3
Registered: 25-1-2018
Member Is Offline
Mood: No Mood
|
|
According dehydratation of HOx.2H2O, seems it is quite easy.
7 g of dihydrate gave me in 3-4 hours almost exactly 5g. There were observed no fumes neiher oxalic acid condensate on the walls. Temperature was
100C, later slowly down to 92C while controlled by weight of product.
In new version of TA purification:
5g of TA (82% purity)+6 mL MeOH
5g of dehydrated HOx + 8 mL MeOH
after mixing (...and serious heat generation), -20C freezer/weeknd, filtration, rinse of few mL of cold MeOH gave about 5.5 g of TAOx. But not yet
complete dry.
So, seems it will have good impact to yield.
[Edited on 29-1-2018 by joe_chem]
[Edited on 29-1-2018 by joe_chem]
|
|
|
Mush
National Hazard
Posts: 633
Registered: 27-12-2008
Member Is Offline
Mood: No Mood
|
|
Very well done joe_chem! 
|
|
|
bipolar
Harmless
Posts: 24
Registered: 24-3-2019
Member Is Offline
|
|
Tryptamine is trolling?
Half a year ago I was making tryptamine by decarboxylation of tryptophan in acetophenone (at 150-180° bath temp).
Tryptamine was isolated and purified via oxalate salt (~50% yield).
Since melting point of obtained product was at 103-105°C, I decided to do futher purification by extraction with boiling hexanes.
It gave product that has a look of absolutely colorless crystals but it still melts at around 105°!
$#@#$?!1! 
I am very confused as the literature gives melting point for tryptamine in ranges from 111 to 118°. Definitely not 105°C.
And I have no idea how to purify this obtained product further.
Recrystallization from MTBE and various aromatic hydrocarbons didn't helped with melting point at all. And vacuum distillation is not an option for me
as I have no access to good vacuum pump.
[Edited on 29-3-2019 by bipolar]
|
|
|
Pumukli
National Hazard
Posts: 705
Registered: 2-3-2014
Location: EU
Member Is Offline
Mood: No Mood
|
|
Bipolar! I'd try sublimation in this case.
I tried sublimating various compounds a while ago and I was pleasantly surprised how useful this process was. My crude setup included a (glass)
Petri-dish placed on a hotplate and a small metal bowl of ice/water placed on the top.
Warning: heat up the plate SLOWLY otherwise the bottom Petri-dish will crack! Done that (several times, just to be sure, you know) :-)
You can check the melting point of the sublimate and for this you can do this on a very small scale.
I found an interesting improvement for the method too: you start with either two Petri-bottoms or two tops and put a pre-perforated filter paper
between these two halves. It serves as a barrier to prevent precious sublimate from falling back to the lower plate when you open the makeshift
"sublimator device". The two halves have to be assembled like this: hotpalte - glass bottom (or top) facing with its rim upwards - perforated filter
paper - glass bottom (or top) facing with rim downward. It requires a steady hand to assemble this structure as the rim against the other rim tries to
slip sideways, especially when you put the ice/water bowl on top. But it can be done. I made about 1-2 mm holes into the filter paper, around 100 such
holes for a 90 mm Petri-dish sublimator.
|
|
|
| Pages:
1
..
5
6
7 |
|








