| Pages:
1
..
48
49
50
51
52
..
66 |
blogfast25
International Hazard

Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by elementcollector1  | | You know, I wonder what it would do to put a piece of sodium in a glass tube or sep funnel at the top of the condenser, to see if it really
is atmospheric H2O or O2 that's destroying my K. I have plenty of sodium. This probably won't be my production method, but it would be another
interesting test (also it would be interesting to see what the sodium metal does). Now, how can I make some sodium wire? Would a plastic pipette be
strong enough, or would the sodium need to be softened first? |
I think you should try and get the temperature right first. When you've eliminated the water as H2, your flask will have been somewhat purged of air
(oxygen) because of the H2 flux. And when you have a nice steady simmer you're also replacing air with solvent vapour...
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Going to see if I can borrow a hotplate from the club, then. (I borrow a lot of things, don't I?)
On a side note, 50 pages! Is this the longest running SciMad thread?
EDIT: Trial 7 is running as we speak. Definitely over 200 C, the solvent appears to be boiling (or is that hydrogen?) and things seem to be going
smoothly. No visible potassium, though...
[Edited on 15-3-2013 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
What in the name of Feynman?
My solvent's turning brown...
EDIT: And now I am out of available tert-butanol. Fantastic.
[Edited on 15-3-2013 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Welp. Success?
Changes:
-Used Pok's masses and such instead of NurdRage's (same as Run 6)
-Corning hotplate w/stirring, borrowed from Science Club
I think what happened is this: Potassium metal was formed, but the alcohol left solution (it kept freezing just above, I had to push it back in with a
metal rod) or got consumed in some side reaction, and the potassium reacted with oxygen to form a brownish-black potassium oxide. HOWEVER! I have
proof that the metal was formed at some point.
When I dumped out the contents into a beaker to inspect them, some was left behind in the flask. I decided (foolishly) to clean this out with water.
There was a hiss at first, then a bright flare of lilac and a pop!
I think there are very small K metal pieces dispersed in this opaque solution of potassium oxide. But how to collect them? My best guess would be to
heat the beaker to the melting point of potassium (about 65 C) and turn on slow stirring, without a stirbar. The potassium, being paramagnetic, should
collect in the center and coalesce, forming a larger and possibly more easily removed sphere.
In the meantime... What do I do with all this potassium oxide? I have the beaker under plastic wrap, in a halfhearted attempt to prevent too much air
access (the solvent well do well enough as it is for a short while). Is there any way I could extract it, or turn it back to potassium?
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
EC1:
Forget about the potassium oxide, it's basically irrecoverable, IMHO and not worth trying.
But clearly you're very nearly there! Too vigorous cooling appears (again) to be your problem: without or with very little t-butanol in the flask, the
reaction speed MUST suffer (that is at least one thing we can safely conclude from the proposed reaction scheme). The fact that at least SOME K formed
when much of your catalyst wasn't even playing is a good sign. I'm now wondering is some of the other failures we've seen from other experimenters may
also be due to freezing t-butanol. You might still be a trailblazer!
MIMP:
I've had solvent turn brown too but it didn't impeded K being formed.
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
So, how to solve this? The water in the fume hood is bound to be a little bit warmer... Maybe...
But is there a way to warm the water up so that it won't freeze the t-butanol, but can still recondense it?
Oh, right. Repoured stuff into the flask, and there were many tiny but bright specks of light, likely more potassium. Doesn't look like anything big,
but as soon as I get home I'll give coalescence a shot.
[Edited on 15-3-2013 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
With a BP of only 83 C, the t-butanol will always condense fully, so no worries there. To avoid actual freezing up, the internal wall of your
condenser should never be below 25 C. Bear in mind that your boiling solvent will help heat up the condensing channel.
I'd go with air cooling: just a bit of air from an aquarium pump, pumped in from the top inlet. Alternatively, try first with no air at all: just
monitor the top of the condensate (aka the 'condensation ring') in the condenser, making sure it doesn't climb too high (that's when loss of vapours
could occur).
It's quite interesting that many of us used nothing more than a glass pipe with some moist kitchen towel and didn't suffer these frozen t-butanol
problems. Sometimes simples does it well...
[Edited on 15-3-2013 by blogfast25]
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Hmm. I know a friend who has an aquarium pump, I'll ask him.
EDIT: 3 whole milliliters of t-butanol were hiding from me! Bad alcohol!
[Edited on 16-3-2013 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Well, good luck. My little finger tells me you're about to make significant balls of potassium.
When I think about it logically, I think too low a temperature (of the plate/flask) goes a long way to explain your fails. Below BP there isn’t much
solvent vapour in the head space of the flask but the vapours nonetheless contain much t-butanol, due to its low BP and high vapour pressure at >
160. The too cool (helped by little condensation of solvent, which would otherwise heat up the condenser inner surface quite a bit) condenser surface
then condenses the t-butanol, unfortunately freezing it in the process. A downward flow of solvent condensate would also have helped redissolving any
solidified t-butanol but there wasn't much of such a flow. Too low reaction temperature combined with subnormal concentration of t-butanol = no K. It
all fits rather well…
[Edited on 16-3-2013 by blogfast25]
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Your little finger talks to you? 
I'm going to see if I can get adequate cooling with a condenser filled with non-moving water. As the reaction progresses, the water will get steadily
more heated, but will never reach 100 C (although it can still reach 85, causing the butanol to boil off). In that case, I'll probably flow a bit more
water into there, and shut it off again.
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by elementcollector1 | Your little finger talks to you?
I'm going to see if I can get adequate cooling with a condenser filled with non-moving water. As the reaction progresses, the water will get steadily
more heated, but will never reach 100 C (although it can still reach 85, causing the butanol to boil off). In that case, I'll probably flow a bit more
water into there, and shut it off again. |
Good idea. Make sure to monitor temperature, if only by hand: 4 h is a long time and there's updraft from the sand bath too as an additional heat
input to the mante water...
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Well, the sand bath is at least 200 C (probably well over that), the coolant flow is as low as possible (no frozen alcohol visible), and we're 2 hours
in. Stirring is at the lowest possible setting. No K visible....
EDIT: Pictures! >

The general setup. Hot plate's on 3, stirring's on 3... if that means anything to anyone.

The mush in the reaction flask. Not much happening, apart from a crust on the top of the oil.

Well, no needle-like crystals visible. I guess the t-butanol is working, then?
[Edited on 16-3-2013 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
4 hours in: No visible potassium, and all the magnesium is pretty much still there. And still shiny. Ugh... Maybe #7 was just luck? The only
difference is that I didn't add all the catalyst at once, instead doing the 'staggered addition' thing. I guess I'll go back to what I was doing
before, but after 8 different tests, this is just getting disappointing. Going to continue reflux for a few hours more, in case a scientific miracle
happens.
EDIT: I leave this thing alone for an hour and it turns brown again. Well, it's better than nothing. I did turn up the heat and the stirring at the
end, so I guess that's a point for next time. The hot plate surface is oddly discolored slightly yellow, so that's weird. Anyway, I'm expecting there
to be small bits of potassium in there, which I'll never get to coalesce, and the majority of the potassium is now the oxide.
So, notes for next experiment:
-bring up the heat more at the beginning
-add the catalyst all at once - again
-More stirring?
-Running out of KOH now. I probably have enough for the next two runs, but not much after that.
You know, I noticed that a significant portion of Mg remained unreacted in both of the last two runs (kind of assuming on this one, since I haven't
examined the gunk yet). Some of it obviously reacted, but not all of it. Also, there is still a significant quantity of t-butanol in the mix, as it
spewed white gas whenever I put it on a heated surface without cooling (fortunately, it still recondensed).
[Edited on 17-3-2013 by elementcollector1]
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
Well, call me a liar!
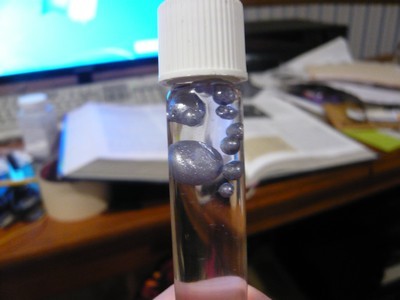
That represents 0.8g of pure potassium, or a theoretical yield of 18.8%. Pretty bad, but it's something. What's the normal yield for this procedure?
Anyway, the reaction mix had turned brown again, so I assumed no potassium was present. I dumped it out into a strainer, and boy, was I wrong.
Oh, and then stuff lit on fire.
NurdRage told me to clean anything that handled potassium metal with alcohol. I did, and it lit on fire anyway. Melted one of my best plastic
containers too. Well, long story short, a towel got burned, the strainer got covered in potassium hydroxide and oxide (thank science I saved most of
the potassium in a beaker with fresh solvent first), and nothing else was recoverable afterwards.
DO NOT USE ISOPROPANOL TO CLEAN POTASSIUM METAL. I used 99% by volume from the local pharmacy, and it caught fire anyways. Then the lamp oil got mixed
in, and that didn't help much. Well, nothing important caught on fire, so nothing harmed too badly.
Well, anyway, after this little fiasco I cleaned and coalesced the rest of the potassium, but quite a number of 1mm spheres of the stuff are left in
the beaker. I can't seem to melt or coalesce these, and I'm frankly too scared to get rid of them with isopropanol. Anything I can do to safely
destroy them?
I put the potassium metal spheres, once they had cooled, into a test tube with fresh mineral oil - good enough for a while. Any tips on ampouling for
a longer storage?
By the way, this stuff is SOFT - even when it was solid and cold, the tweezers used to scoop it out into the test tube severely dented the surface. I
mean, that's to be expected - sodium can be cut with a knife. But still, it's such an interesting property for a metal to be soft.
Elementcollector1, signing off - it's been a long run after 8 tries, but it was worth it! And then I have to do it again in front of my peers.
...Monologue over.
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
mr.crow
National Hazard
Posts: 884
Registered: 9-9-2009
Location: Canada
Member Is Offline
Mood: 0xFF
|
|
Congrats!!!
Try using an inert solvent and adding the isopropanol very slowly. You could also use your tert-butanol
Yes, its been a long thread 
Double, double toil and trouble; Fire burn, and caldron bubble
|
|
|
condennnsa
Hazard to Others
Posts: 217
Registered: 20-4-2010
Location: Romania
Member Is Offline
Mood: No Mood
|
|
congratulations, elementcollector, your success is encouraging to me I plan to try this again soon
|
|
|
Mailinmypocket
International Hazard
Posts: 1351
Registered: 12-5-2011
Member Is Offline
Mood: No Mood
|
|
Well you son of a....!
Lol  very nice though, good job! Ill have to read over your last couple posts in
detail when I have time, but this is very promising! very nice though, good job! Ill have to read over your last couple posts in
detail when I have time, but this is very promising!
K-ongratulations! 
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
EC1: there you go; welcome to the weird world of K! (My little finger didn't lie after all!)
A few points.
Stirring: if your solvent was slowly simmering then a very slow speed of stirring should be sufficient. I never used magnetic stirring at all. Beware
that molten K can react with Teflon (stirrer bars coating) according to:
2n K+ (CF2)n [Teflon] === > 2n KF + n C (potentially very energetically!) Check your stirrer bar for black/grey spots/streaks!
Yield: 50 % or more is easily achieved. A couple more runs and you’re probably there.
Disposal of K: reaction with alcohols like IPA or methylated spirits is much less vigorous than with water. I’m very surprised in your case the K
caught fire with IPA, was the K still warm by any chance?
So be careful with admonitions like: "DO NOT USE ISOPROPANOL TO CLEAN POTASSIUM METAL": many of use use IPA for refreshing the surface of potassium
balls, PRECISELY because it reacts with K only quite slowly.
Ampouling K: we’ll keep that for another day. For now, storing under clean oil is enough.
Heat: apply full heat right from t = 0.
Beware: from your 3rd photo on 16/03 I can see that your bottom water outlet tube was attached to the condenser in quite a dangerous
way. If that hose flips off you've got cold water hitting a hot flask: thermal shock, possibly cracking of the flask and hot K to boot!!!!
[Edited on 17-3-2013 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
And a quick word on yield.
Theoretically achievable yield is less than what most here calculate. Firstly, your KOH contains about 10 % water, so you need to subtract that from
the charge to get the actual amount of pure KOH available for reaction.
Secondly, even if your reaction proceeded ‘100 %’, an amount of K that’s stoichiometrically equivalent to the amount of t-butanol used is
‘locked’ away as K t-butoxide at the end of the reaction. That’s a simple consequence of how this thing works. So if you used 0.1 mol t-butanol,
0.1 mol of K is ‘lost’ because it remains tied up as K t-BuO.
Any calculation of so called Actual Yield must take these realities into account.
|
|
|
m1tanker78
National Hazard
Posts: 685
Registered: 5-1-2011
Member Is Offline
Mood: No Mood
|
|
EC1: That's the prize of stubbornness. Good job!
Note that you can use brake fluid to destroy remaining K bits in the reaction flask without so much drama. Add it then loosely cap the flask in case
any of the bits float up. Swirl occasionally if needed.
Tank
Chemical CURIOSITY KILLED THE CATalyst.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Tank:
I remember trying to coalesce K with DOT and that the K reacted with the brake fluid, albeit very slowly. Presumably K attacks the oxygen in the
polyethers of the DOT. But these atoms are much more sluggish than the -OH groups in alcohols. Ethanol, propanol etc all react well with K/Na without
much drama.
|
|
|
m1tanker78
National Hazard
Posts: 685
Registered: 5-1-2011
Member Is Offline
Mood: No Mood
|
|
Blog: I do recall your experiment. EC1 claimed that his mixture caught fire when he added IPA. I suggested brake fluid as a low or no drama solution
but you're right, alcohols will react much more vigorously. Methanol would probably be the best since absolute IPA and ethanol aren't always OTC.
To paraphrase EC, "little fiasco" = he wasn't prepared for the fire but luckily no harm done.
Tank
Chemical CURIOSITY KILLED THE CATalyst.
|
|
|
elementcollector1
International Hazard
Posts: 2684
Registered: 28-12-2011
Location: The Known Universe
Member Is Offline
Mood: Molten
|
|
I think the K was still quite warm, as I remember having trouble getting the solution down to room temperature.
Oddly enough, the stirbar did react, but only in the middle, forming a yellowish-gray coating. Wiped off easily, so no worries there.
Anyway, I guess I'll try alcohol to destroy the bunch of little bits of K, but I'm going to do it outside this time.
What I had done was I got a metal strainer and a plastic container, and strained the mix through (here I first saw the odd, lumpy bits of K). I
scooped these out into a beaker of fresh lamp oil, and some smaller bits of K had fallen through the strainer (without my knowledge). I poured
isopropanol over the mix in a futile attempt to clean the strainer (too big to submerse in alcohol, but couldn't hold any of the liquid either), and
several small bits of K rose to the surface, quickly reacting. And then they caught fire. I suppose it was a combination of the alcohol, kerosene, and
warm K metal that did it, but still, the fire was for the most part contained. I took a metal spoon, and scooped the flaming plastic container into a
larger metal one, setting that on the floor and placing the steel pot with the sand bath over it (this cut off oxygen, and extinguished the flames
handily). Now, some bits of flaming material had also made it onto a plastic bag and pink towel on the floor, and these caught fire too. I stamped
those fires out, and proceeded to throw the towel, strainer and bucket with the now-charred mix outside, where it was heavily raining. I thought I saw
a tiny droplet of K leap out of the metal container, hit the ground, and produce a tiny lilac flame, but it was quickly extinguished. After this, I
kept the garage door open to reduce the smell of burnt plastic, and focused on coalescing the K.
We'll be back with 'Misadventures in Chemistry' every Sunday morning, stay tuned!
I guess I'll use alcohol, but I'm probably going to pour off the lamp oil first, to avoid too much kerosene in the mix.
That brings me to an important note - when I was coalescing the K in fresh lamp oil, this yellowish fluff started to appear, turning the beaker
cloudy. What could this be?
Elements Collected:52/87
Latest Acquired: Cl
Next in Line: Nd
|
|
|
m1tanker78
National Hazard
Posts: 685
Registered: 5-1-2011
Member Is Offline
Mood: No Mood
|
|
Impurities in the lamp oil, most likely. I always get some sort of grunge when I coalesce sodium or NaK. What brand is your lamp oil? If you're in the
U.S., the best lamp oil I've used so far is from Academy. Here is a link to the product.
Tank
Chemical CURIOSITY KILLED THE CATalyst.
|
|
|
Lambda-Eyde
National Hazard
Posts: 860
Registered: 20-11-2008
Location: Norway
Member Is Offline
Mood: Cleaved
|
|
Congratulations, ec1. I have run this reaction once and I didn't get any potassium either. I used paraffin oil as the solvent, Merck t-BuOH, old KOH
(which was >85,5% a few decades ago) and a freshly opened bottle of Mg turnings. I performed the reaction in a 250 erlenmeyer and used a 300 mm
vigreux as a condenser. A bubbler was attached to the vigreux. I can't say what the temperature was for sure, my thermocouple gave very different
reading depending on the placement of the probe. It peaked at around 370 C (!) in some positions, but the sand bath seemed to be stable at 220-250 C.
Whether or not my probe is faulty or if this is a typical reading I don't know. There was a very visible reaction between the KOH and the Mg when the
reaction reached a certain temperature. The vigreux was lukewarm through the whole procedure. I can't remember how I added the catalyst. I will come
back with a more detailed report when I get home to my notebook. I will try the experiment again as well.
This just in: 95,5 % of the world population lives outside the USA
Please drop by our IRC channel: #sciencemadness @ irc.efnet.org
|
|
|
| Pages:
1
..
48
49
50
51
52
..
66 |








