| Pages:
1
2
3
4
5
6 |
Jimmymajesty
Hazard to Others

Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
One of the pdf that S.C. Whack uploaded mentioned that during their experiements, an explosion occured when fuming nitric acid was used!
I thought that the dissolved NOx will be readily swept out by means of the hydrocarbon feed, so my plan was to use ~98w% red nitric, and bubble
lighter filler gas through it, without heating the bubbler, until the nitric becomes colourless.
I poured ~250ml >98% HNO3 into a 1l bubbler, and started to feed hydrocarbon into it, after about 30min the nitric became hot to the touch (damn)
and a lot of brown fumes come out of it, after one hour the bubbler was stilll hot. Unfortunately the dissolved NOx cannot be swept out so easily as I
thought, so it cannot be used as it is.
The dissolved NOx oxidize the butane and heat the nitric as a result of that, more NOx forms which oxidizes more butane, so one will definitely ends
up in hospital if use fuming nitric to GP nitration, hope I saved some life with this post, mine included
Based on my small scale experiments, I found that the vapour pressure of the cc nitric is high enough to make a HNO3 hydrocarbon mix by simply bubble
the gas through the acid, and also the bursting bubbles that come up to the surface will volatilize more nitric which ensures more HNO3 in the vapour
phase by which I managed to react the CH.
Now cc HNO3 cannot be used, and diluted HNO3 is sucks too because of the aforementioned. I will try to decolorize the nitric with a lil urea and pass
hydrocarbon through it to see if it warms.
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
The nitric acid warmed after urea treatement as I'd expected, so I poured about 150ml cc H2SO4 to 100ml ccHNO3 and bubbled the CH into the mixture.
Fortunately there was not any NOx produced as can be seen on the photos.. the brown was taken from pure nitric + CH, the colourless was taken from the
H2SO4+HNO3 mixture, the acid mixture only slightly warmed when CH was being passed through it, so I will definitely continue the experiments with
that.
Attachment: CH HNO3 mixer.doc (236kB)
This file has been downloaded 1052 times
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
When I was doing this experiment, I got an aldehyde smell when I turned up the temperature too high. Typically, it was a combination of formaldehyde
and acetaldehyde. My glass tubing was either 4mm or 5mm in diameter, and I never got any violent reactions. A few times I saw jets of flame shoot
through it when nitric acid would condense in the tube leading from the nitric acid bottle to the reaction tube. That would cause some liquid nitric
acid to get in the reaction tube and make the flame jet.
Are you sending the nitric acid/butane mixture through a 1mm tube at any point? That could make it explode due to a buildup in pressure and the
higher boiling point of butane. Propane seems like it'd be a bit safer as it would have less tendency to be in a liquid phase outside of its
container.
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
You can avoid the small explosions by sloping the initial part of the tubing in such a way, that any condensed nitric will flow back to the bubbler. I
do not think personally that is represents any danger.
As for the small diameter of the quartz tube, see attached photo I think that was
the study of gas phase nitration on smallest scale ever, the bubbler capacity is less than 5ml, I make it explode many times, and it only made sound
but did not crack.
I am currently working on implementing the cooling and protection apparatus outside of my lab, I prepare to the worst, 250ml nitric is not something
that I would fuck around with carelessly.
Unfortunately lighter filler gas is my only sourse of hydrocarbons, except for some other exotics
Attachment: small scale.doc (89kB)
This file has been downloaded 1135 times
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
You don't have those little propane tanks for blowtorches where you are? If you have a slightly larger diameter tube, it'd probably be safer to use
that, since you'd be less likely to get a pressure buildup behind the tube. That narrow tube might also have too much surface area. The literature
did say a high surface area to volume ratio could cause oxidation instead of nitration, although that may depend more on the tube material.
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
I am going to use d=1cm glass tubing. I asked the glassblower to make it from such a long tube that eventually the tubing capacity would be about
200-250ml. I have not checked its capacity yet, and I think it does not matter, because you can adjust the temperature and the flow rate anyway. And
literature suggests 0,1-20sec residence time, which means it is not that important.
I am going to adjust the temperature first, and when I get some product, continue with the flow rate.
Propane tank for blow torches are mainly contains the same gas mix, I checked it, no pure propane. but I also does not worry about that, as my goal is
hydroxylamine.
Seemingly there is not much thing I worry about, there is only one, the yield 
I foreseen that, after a number of cans I will only get 10-20ml impure product
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Substrates other than alkanes can also give nitroalkanes upon radical nitration in the gas phase. For example, nitration of acetone with NO2 in the
gas phase gives a mixture containing nitromethane as the only nitro compound and acetic acid as the main product (see US4517393). Even though the
yield is poor, it should be safer to run than nitrations using HNO3 on alkanes, though it might not work well at 1 atm.
Furthermore, even alcohols, ethers and carboxylic acids can be nitrated in the gas phase to give nitroalkanes in most cases (see JACS, 76, 2692–2694 and references therein).
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
The acetone one would be nice. I am playing with the idea ever since took up this project of using alcohol, acetone and the like as a carbon source,
but I am in short of image of workable setups.
My idea would be to use an Y element, one arm would be for the alcohol, and another for nitric.
Nitric acid would simply be heated by a mantle, and the acohol would be evaporated by dripping it into a heated quartz tube. Problems that I can see
are that the nitric acid would be kept near its boiling point for hours, so a lot of NOx would be generated instead of HNO3 vapour.
Also the flow rate should be relatively fast at each feed to keep a constant mole ratio, so the reactants wouldnt spend much time in the hot tube
which means lower reaction temp.
To keep a constant mass flow by dripping something, is also problematic.
The mixing of the two reactants at the initial stage of the hot tube would also be probably inadequate. I fear that without mixing I simply burn the
nitric then pass through alcohol and water vapour.
Do you guys have some nice idea that would eliminate all these problems?
|
|
|
watson.fawkes
International Hazard
Posts: 2793
Registered: 16-8-2008
Member Is Offline
Mood: No Mood
|
|
For constant reagent rates through
a drip orifice, use peristaltic pumps with variable rate drives. This will get you close enough in ratio.
For a reaction chamber, use a long, vertical tube. Introduce alcohol vapor at the bottom and have a gas takeoff on the top that contains unreacted
alcohol and all the reaction products. Introduce nitric acid through a spray nozzle at the top in very fine droplets. Collect unreacted liquid acid at
the bottom of the tube. You'll might want a condenser or cold finger in the dead space below the inlet to reduce NOx vapor pressure.
I've addressed the mixing issue, in part, with the counter-current flows that this apparatus has. The reaction tube should be, at the least, insulated
and probably also heated. Standard glass tube lengths from the factory are 120 cm, or about 4 feet, and I'd start with a single full length. You could
use two of them in a room with high ceilings before you need to consider special building construction.
|
|
|
Jimmymajesty
Hazard to Others
Posts: 153
Registered: 9-7-2009
Member Is Offline
Mood: No Mood
|
|
Before the large scale test I also conducted a small scale test with the H2SO4+HNO3 mix, it worked equally well again HCHO and a peculiar smell at
some point.
I measured the bubbler and the gas can weight and measured them after 15 and 45 min of operation. Based on the results the CH/HNO3 mole ratio is well
above infinite.. 59g propane butane mixture vaporized only one g HNO3, after the bubbler continuous HNO3 fumes were escaping though. After these
experiments, I assembled the setup that can be seen in the attachment. (everything was grounded glass jointed except the connection between the two
condensers).
The reactor capacity was about 200ml d=1cm. The furnace was made of two hotplates welded opposite to each other (each hotplate wattage was 1000W)
and temperature controlled. The first condenser was a spiral type the second an allihn type, the first only cooled the gas mixture the second
condensed back any vaporized stuff.
I poured ~150ml cc HNO3 and 150ml cc H2SO4 into the bubbler and waited till it cooled.
I started to heat up the furnace and at about 100°C I attached a CH can to the end, to flush the air out of the setup.
Then I attached the can to the front part and slightly opened it, waited till the temp of the furnace reached 380°C, at 250°C the brown fumes after
the reactor tube disappeared, so probalby the flow rate was at its optimum at that temperature. At 380°C I could smell the carrot like smell again,
which I think the pyrolysis products of the nitroparaffin, so I set back the SP of the controller to 350°C and adjusted the CH flow in such a way,
that the brown fumes had just disappeared. At this point a peculiar smell could be noticed at the outlet of the second condenser, It was like
isopropyl nitrite, a condensate is started to drip into the flask of the first condenser, with a rate about one drop/25 sec. The condensate separated
into two phase, but they reacted with each other since the bottom of the flask warmed. This was probably due to the formed HCHO and HNO3 made it at
the heating up period into the first receiver flask.
After about 30min I sucked up the condensate from the first flask with a syringe, then washed with water, and smelled, its smell was between menthol
and nitro toluene, or rather between paraldehyde and nitro toluene, it was a pleasant smell.
In the second receiver flask only oxidized organic stuff was present which were completely dissolved upon addition of water.
I lit a couple of drops with open flame and it burned with a nice grey colour.
I do not recommend to reproduce this experiment, since it is low yielding, especially at these circumstances. It was only good for get some product to
smell it.. I did not heat the bubbler though, by which I think I could attain better results. The explosion hazard problem should be solved by
lowering the bubbler head space to zero without HNO3 droplets being sprayed into the hot tube, any ideas on this?
BTW the HNO3+H2SO4 mixture, also heated on its own, it was at about 35°C at the end of the experiment, also I do not think that the nitric could be
regenerated from the mixture, I suscpect that there is a lot of semi oxidised CH crap in the mixture that would cause serious frothing upon heating.
Watson interesting approach I tried that too, but how can you make an even spray,
that will be volatilized before falling into the high temp CH vapours? Its seems to be a problem that only industrial bastards can solve over a
designer table, I took a 1m long quartz tube and wounded some kanthal wire to the bottom, then I heated the bottom and slowly flooded the bottom of
the tube, in such a way that a reflux of CH developed, (I could only hold this state for a min though, then overflow, etc.) during that short time I
tried to drip, spray, pour HNO3 into the tube but this always resulted in a cracking noise & barking dog sound..
Attachment: GP nitration exp setup.doc (265kB)
This file has been downloaded 1030 times
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Considering that the industrial methods usually employed are pretty tough to conduct with home made stuff... Once again, I might consider some
alternative.
IF Shulgins procedure for de-constructing the B-Nitro-propene to produce Myristicin Aldehyde and Nitroethane is applicable. Then, the nitration of
styrene with I2/NaNo2, in ethylene glycol, to produce B-Nitrostyrene....might be a good starting point, to produce Nitromethane and Benzaldehyde.
I can easily and cheaply obtain all of the ingredients, and solvents. And, the yield of Beta-Nitrostyrene is purported to be about 80%.
With luck, refluxing the resultant B-Nitro-Styrene with N-Methyl Benzylamine will produce the Benzaldehyde-imine.....Plus very pure Nitromethane as
gas.
High grade Nitromethane, and an equivalent amount of Benzaldehyde should be recovered (After performing hydrolysis on the imine). I can also recover
and recycle my Iodine and N-Methyl-Benzylamine.
Likewise, if this Nitration could be performed successfully on Iso-Eugenol , The deconstructed Nitropropene would yield Vanillin and Nitroethane.
I'm not sure if this nitration can be performed on Iso-Eugenol.....Maybe not.
http://www.erowid.org/archive/rhodium/chemistry/nitryliodide...
[Edited on 12-7-2010 by zed]
[Edited on 12-7-2010 by zed]
[Edited on 12-7-2010 by zed]
|
|
|
madcedar
Hazard to Others
Posts: 116
Registered: 10-9-2009
Member Is Offline
Mood: No Mood
|
|
Nitroethane from ethanol and nitric acid
Since you are in the mood for experimentation here is a patent for making nitroethane from ethanol and nitric acid (and other useful information).

Attachment: nitroethane pat4431842 (ethanol and nitric acid).pdf (257kB)
This file has been downloaded 2253 times
Attachment: Nitroethane, Properties and Azeotropes.pdf (139kB)
This file has been downloaded 1950 times
Attachment: Nitroethane, Purification by Azeotropic Distillation US3480517.pdf (118kB)
This file has been downloaded 1186 times
|
|
|
arsphenamine
Hazard to Others
Posts: 236
Registered: 12-8-2010
Location: I smell horses, Maryland, USA
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by zed  | | IF Shulgins procedure for de-constructing the B-Nitro-propene to produce Myristicin Aldehyde and Nitroethane is applicable. Then, the nitration of
styrene with I2/NaNo2, in ethylene glycol, to produce B-Nitrostyrene....might be a good starting point, to produce Nitromethane and Benzaldehyde.
|
Maybe I'm too easily amused, but the thought of a "reverse Henry Reaction" is chortle producing.
Here someone has a perfectly good illicit precursor and they want to disproportionate it back into Henry reactants.
|
|
|
stygian
Hazard to Others
Posts: 242
Registered: 19-9-2004
Member Is Offline
Mood: No Mood
|
|
It's a more interesting idea if you could make it work with 2-nitropropene.. and fancied working with 2-nitropropene
|
|
|
zed
International Hazard
Posts: 2283
Registered: 6-9-2008
Location: Great State of Jefferson, City of Portland
Member Is Offline
Mood: Semi-repentant Sith Lord
|
|
Ummmm. Well, P-Propenyl anisole and Iso-Eugenol, are inexpensive and fairly worthless as precursors.
Using them to generate, difficult to obtain Nitroethane, ala Shulgin....has some merit.
Additionally , Vanillin and Anisaldehyde, have their own uses.
Shulgin ripping apart the MMDA skeleton to produce Myristicin Aldehyde, may seem
crazy to us, but it suited his goal. And, it served to reveal a potential Nitroethane synthesis, that could prove very handy in this era when
Nitroethane is very tough to come by.
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
| Quote: |
Nitromethane can be prepared by the action of dimethyl sulfate on potassium nitrite
Walden, Ber. 40, 3216 (1907)
|
http://www.orgsyn.org/orgsyn/prep.asp?prep=cv1p0401
Anyone know detail of this reaction?
|
|
|
franklyn
International Hazard
Posts: 3026
Registered: 30-5-2006
Location: Da Big Apple
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Waffles SS | Nitromethane can be prepared by the action of dimethyl sulfate on potassium nitrite
Walden, Ber. 40, 3216 (1907)
Anyone know detail of this reaction? |
read second half here
http://www.sciencemadness.org/talk/viewthread.php?tid=13633&...
A good reason not to make that way
http://www.sciencemadness.org/talk/viewthread.php?tid=1608&a...
http://www.sciencemadness.org/talk/viewthread.php?tid=1608
http://www.sciencemadness.org/talk/viewthread.php?tid=2995
http://www.sciencemadness.org/talk/viewthread.php?tid=9570
.
|
|
|
MeSynth
Hazard to Others
Posts: 107
Registered: 29-7-2011
Member Is Offline
Mood: http://www.youtube.com/watch?v=5ZltqlVuDIo
|
|
| Quote: Originally posted by 497 | This patent might be interesting to some here. It is a patent that gives the conditions to produce a mixture of 83% nitroethane and 17%
nitromethane by vapor phase nitration of propyl nitrite with 50% HNO3 vapor at 400*C (or NO2 at 300*C), at atmospheric pressure. Interestingly, they
use an aluminum tube immersed in a molten salt as the reactor. It is advantageous because it results in a simpler mixture of products
that should be much easier to purify... Assuming you can get n-propyl alcohol, propyl nitrite is ultra easy to synthesize.. The extra carbon is lost
as formaldehyde. If you wanted nice pure nitromethane, you could use isopropyl nitrite instead, which results in only nitromethane (and acetaldehyde
byproduct).
[Edited on 18-6-2010 by 497] |
US3209038
I have tried to work out how this would be done. Saftey is a major issue due to n-propyl nitrite being an explosive. Due to the explosive nature of
the reactants I would not hand operate this and would rather operate the air pump from behind a concrete wall. Other than this explosion hazard I feel
no threat and have drawn out the first part and the second leaving out the n-propyl nitrite stage since I'm not certain about what they mean in the
patent when they say the n-propyl nitrite was allowed to enter a stream of 50% nitric acid vapor that has been preheated to 260* C. The pictures have
been added as attachments.
My best guess is small amounts of n-propyl nitrite will boil out when the nitric acid vapor heated to 260* C. is passed through causing just the right
amount to vaporize and mix with the gas.
The missing stage is where the nitric acid combines with the n-propyl nitrite before entering the reactor.
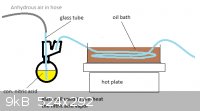 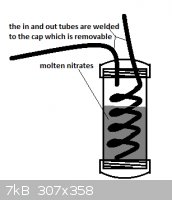
|
|
|
497
National Hazard
Posts: 778
Registered: 6-10-2007
Member Is Offline
Mood: HSbF6
|
|
PrONO has a terrible oxygen balance on its own, it would be tough if not impossible to detonate in my opinion. Mixing it with HNO3 would improve the
OB and sensitivity no doubt, but it shouldn't be hard to avoid that fully by using an inert gas to dilute the gas mixture. Concrete walls are always a
good precaution though if you have them. It shouldn't much need hands on attention anyway.
I'm not certain, but I would bet that the mixture of HNO3, H2O and RONO proposed in the patent forms an equilibrium with ROH, NO, and NO2 via 2RONO +
H2O <---> 2ROH + NO + NO2.
Since NO2 can be formed from air by electric discharge pretty easily, you might be able to have a continous process where the output from the electric
discharge is fed into an absorption column/bubbler filled with liquid ROH + H2O. These react to form RONO + HNO3, at least until too much HNO3 builds
up. Then the ROH + RONO + HNO3 + H2O from the NO2 absorber is dripped into a vaporizer and piped into you reactor tube. Supplemental HNO3 could also
be added from a separate NO2 absorber filled with only H2O if need be. HNO3 recycled back from the product mixture would likely be sufficient though.
Electricity, air, water, and an alcohol in, nitroalkanes and aldehydes of your choice out. Sound pretty elegant to me. The inert gas could hopefully
be recycled. CO2 is pretty cheap though. I'd be interested in using it for both the acetaldehyde + nitromethane, and the formaldehyde + nitroethane
possibilities.
Not sure about the best ways to process the end mixture of HNO3, ROH, water, nitroalkanes and aldehyde(s). Nitroethane does form azeotropes with
n-propanol and water for sure. Maybe an extractive distillation could help?
What do you think?
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
The vapor phase nitration of propane with nitric acid at 420degC produces various nitroalkanes in 40% yield total.
The yield for nitromethane is only 4%, for nitroethane 10%, for 2-Nitropropane the yield is 16%.
The various nitroalkanes are then separated by distillation. Nitromethane boils off first at 101degC, then nitroethane at 114degC. The vapor-phase
nitration can alternatively be done with nitrogen dioxide, giving better yields, with the additional advantage that the products contain less
2-nitropropane.
2-Nitropropane boils at 120degC. 1-Nitropropane boils at 131degC. 2-Nitropropane is a carcinogen, breathing of its vapor should be avoided.
[Edited on 22-9-2011 by AndersHoveland]
|
|
|
497
National Hazard
Posts: 778
Registered: 6-10-2007
Member Is Offline
Mood: HSbF6
|
|
I don't see how effective separation of of nitromethane, nitroethane, and nitropropanes can be accomplished with their boiling points all being within
30 degrees of each other.. Even worse is that nitroethane and 2-nitropropane are within 6 degrees or each other.. I think a method that produces
nitroethane more selectively would be much more useful. Unless there is a magic bullet way to purify it?
|
|
|
simba
Hazard to Others
Posts: 175
Registered: 20-5-2011
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by 497 | | I don't see how effective separation of of nitromethane, nitroethane, and nitropropanes can be accomplished with their boiling points all being within
30 degrees of each other.. Even worse is that nitroethane and 2-nitropropane are within 6 degrees or each other.. I think a method that produces
nitroethane more selectively would be much more useful. Unless there is a magic bullet way to purify it? |
Maybe they can be purified by freezing...nitromethane freezes at −29 °C, nitroethane at -90 °C, and I don't know about nitropropanes.
|
|
|
AndersHoveland
Hazard to Other Members, due to repeated speculation and posting of untested highly dangerous procedures!
Posts: 1986
Registered: 2-3-2011
Member Is Offline
Mood: No Mood
|
|
It might be better to do a vapor phase nitration using ethane, or possibly even methane (natural gas), to avoid formation of hazardous 2-nitropropane.
Typically, when two similar compounds have very close boiling points, the mixture needs to be distilled several times repeatedly. This can generally
minimize impurities in the separated compounds.
For example, the portion that initially distills off from a mixture of nitromethane and nitroethane would be redistilled again, and the portion that
distills off from the second distillation would be nearly pure nitromethane, although some of the nitromethane got left behind with the nitroethane.
The industrial process only uses propane because nitroethane, which is used as an industrial solvent, actually sells for a higher price than
nitromethane. Nitromethane is considered the byproduct.
[Edited on 22-9-2011 by AndersHoveland]
|
|
|
Alastair
Hazard to Self
Posts: 59
Registered: 13-7-2011
Member Is Offline
Mood: Barely any solvent in my emulsion
|
|
''For example, sodium (nitrite) and potassium nitrite reacting with iodomethane would produce mostly nitromethane, with methyl nitrite as the minor
product''
Source: wikipedia. ref: Donald L. Pavia, Gary M. Lampman, George S. Kriz (2004). Organic Chemistry. 2. Mason, Ohio: Thompson Custom Publishing. ISBN
0030148138. OCLC 236055357 But i couldnt find the ref.
Could this be applied to iodoethane?
Or maybe nitroethane can be separated as a side product from nitrite syntheses via alcohols + NaNO2 (or N2O3) ?
|
|
|
simba
Hazard to Others
Posts: 175
Registered: 20-5-2011
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by Alastair | ''For example, sodium (nitrite) and potassium nitrite reacting with iodomethane would produce mostly nitromethane, with methyl nitrite as the minor
product''
Source: wikipedia. ref: Donald L. Pavia, Gary M. Lampman, George S. Kriz (2004). Organic Chemistry. 2. Mason, Ohio: Thompson Custom Publishing. ISBN
0030148138. OCLC 236055357 But i couldnt find the ref.
Could this be applied to iodoethane?
Or maybe nitroethane can be separated as a side product from nitrite syntheses via alcohols + NaNO2 (or N2O3) ? |
yeah...iodoethane or any other haloethane will yield nitroethane.
|
|
|
| Pages:
1
2
3
4
5
6 |









