| Pages:
1
..
28
29
30
31
32
33 |
blogfast25
International Hazard

Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Take no notice: it was me getting the structure of p-TsOH wrong!
Thanks for pointing it out.
[Edited on 18-1-2016 by blogfast25]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Dear students and faculty members of B&D University,
We are pleased to announce that the teachers' strike resulting from the recent budget cuts has officially concluded. After three long months of
non-stop protesting, the lengthy standoff between B&D University and members of its chemistry department finally came to an end Wednesday evening
after the successful negotiation of a new contract. Under this new agreement, the university will significantly increase all chemistry-related
funding, allowing the school's laboratories to be retrofitted with the modern glassware and equipment necessary to conduct college-level
experimentation. For the first time since its inception, B&D University will no longer force students to carry out their chemical reactions via
makeshift condensers and flasks made from copper pipes and pickle jars.
And now that I'm back from striking, I can't help but wonder how B&D University's brightest (and only) pupil has been doing since I've been away.
Is aga still on his noble journey to learn organic chemistry or has he decided to throw in the towel and go back to inorganic?
[Edited on 3-3-2016 by Darkstar]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
@Darkstar:
Carbanions, Darkstar, carbanions! Organometallics, that's one subject that's been 'studiously' avoided so far...
|
|
|
Magpie
lab constructor
Posts: 5939
Registered: 1-11-2003
Location: USA
Member Is Offline
Mood: Chemistry: the subtle science.
|
|
There's a subject I would like to see B&D university cover: activity coefficients and fugacity. I know I can read about these
on Wiki but what I'm looking for is how a chemist actually uses them.
If you can't look them up of what value are they? Can they be derived?
When are they of value to the home chemist?
The single most important condition for a successful synthesis is good mixing - Nicodem
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by Magpie  | There's a subject I would like to see B&D university cover: activity coefficients and fugacity. I know I can read about these
on Wiki but what I'm looking for is how a chemist actually uses them.
If you can't look them up of what value are they? Can they be derived?
When are they of value to the home chemist? |
Activity coefficients were briefly covered here with some worked examples, in case you missed it.
What use these concepts have to the home scientist is the same for professional chemists: it depends enormously what you're doing or are trying to
achieve.
[Edited on 3-3-2016 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
| Quote: Originally posted by Darkstar | | no longer force students to carry out their chemical reactions via makeshift condensers and flasks made from copper pipes and pickle jars.
|
Yay ! At last something worth stealing from the lab !
| Quote: | | ... wonder how B&D University's brightest (and only) pupil has been doing since I've been away. Is aga still on his noble journey to learn organic
chemistry or has he decided to throw in the towel and go back to inorganic? |
Got stalled in the simple (!) process of making a tertiary alcohol.
Depression set in over the senseless waste of so many innocent reagent's lives, then turned to booze and drugs, as is the B&D University
tradition. Hopefully the new lab equipment will make that cheaper.
Speaking of which, exactly how does a tiny amount of water interfere in the chlorination of ethanol ?
(anhydrous=rapid formation of white trichloroacetaldehyde precipitate, any water=clear, sweet smelling solution).
Glad to see you back Darkstar !
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
Speaking of which, exactly how does a tiny amount of water interfere in the chlorination of ethanol ?
(anhydrous=rapid formation of white trichloroacetaldehyde precipitate, any water=clear, sweet smelling solution).
Glad to see you back Darkstar ! |
Do post that question with relevant data in the t-alcohol thread, old chap. We'll try and tend to it.
'Lectures' will resume very shortly here.
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by aga | Speaking of which, exactly how does a tiny amount of water interfere in the chlorination of ethanol ?
(anhydrous=rapid formation of white trichloroacetaldehyde precipitate, any water=clear, sweet smelling solution). |
This should probably be discussed over in blog's OC thread, but isn't the alcoholate the primary product when chlorinated under anhydrous conditions?
I'm guessing the white precipitate is chloral alcoholate. That would make sense considering the acidic environment due to HCl production and the
excess ethanol present, which would shift the equilibrium in favor of hemiacetal formation. Why the chloral doesn't go all the way to an acetal under
those conditions, I have no idea. Perhaps the hemiacetal is unusually stable for the same reason the hydrate is thanks to the adjacent
electron-withdrawing chloro groups? If you have the means, you could try a melting point test on the precipitate.
Also, I kind of doubt that water is directly interfering with the chlorination since acetaldehyde is commonly chlorinated under aqueous conditions to
give chloral hydrate. Which also leads me to believe that you're probably producing the hydrate when using wet ethanol. I'd imagine that the hydrate's
solubility would be somewhat increased due to the extra water present. And since the hydrate is likely more soluble in the ethanol-water mix than the
chloral alcoholate is in the anhydrous ethanol, that might also explain why there's no precipitate in the case of the former.
Anyway, we should probably discuss this in the other thread. I'm also down with continuing your "education," so just let me know if you'd like to.
Perhaps we can even make a lesson out of this reaction? I see that blogfast has already lightly touched on some possible mechanisms in the OC thread.
I'm thinking this reaction would be a good introduction to bond homolysis, free radicals and free-radical halogenation. And considering the increasing
popularity of using bleach to oxidize alcohols, that initial oxidation of ethanol to acetaldehyde by chlorine makes for a rather interesting
discussion all on its own. My guess is that it goes through an unstable ethyl hypochlorite intermediate that quickly decomposes into acetaldehyde and
HCl.
Speaking of which, I actually made a post a while back about these sorts of oxidations if you're interested. The mechanism proposed was for the oxidation of THF to GBL using
aqueous calcium hypochlorite in acetonitrile with acetic acid as the catalyst. And although the mechanism shows the active oxidizing species to be an
activated hypochloronium ion (H2ClO+), these days I actually tend to lean more towards molecular Cl2 itself as the
real oxidizer in bleach oxidations.
Edit: Now that I think about it, the ethanol in the chloral synthesis could very well also be oxidized via a competing radical disproportionation
mechanism as well, bypassing any sort of ethyl hypochlorite intermediate. Welcome to the wonderful world of radical chemistry where no one has any
real clue as to what the hell is actually going on.
[Edited on 3-7-2016 by Darkstar]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Woohoo !
Some theory on the clorination of ethanol would be great, seeing as that's the first step in this synthesis i'm failing with.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Carbon atoms as Lewis bases:
Recapping from what we seen so far, many reactions in OC can be summarised by the following scheme:
A+:B→A-B
Where A is a Lewis acid, :B a Lewis base and : an unbonded electron pair and A-B the adduct. Of course this step may be preceded and/or followed up
with other steps to make up the full reaction path.
So far, our Lewis acid has invariable been a partly positively charged C-atom or a carbocation.
Is it possible for a C-atom to play the part of a Lewis base, an electron pair donor? Enter organo-metal compounds.
Organo-metal compounds:
For those unfamiliar with this class of chemical compounds, there’s at least one you’ll have heard of, i.e. the now infamous lead-based
anti-knocking agent tetraethyl lead:

A more interesting one is methyl lithium, CH3Li, or LiMe.
Over-simplified (the real structure is more complicated and less than fully ionic), LiMe would be made up of negatively charged CH3
so-called carbanions and positively charged Li cations. The structure of the carbanion is shown below:

The Me carbanion has a tetrahedral structure with an unpaired, filled MO sitting ‘on top’. This makes the ion a very hard Lewis base.
To the right is a generically substituted carbanion. The substitutions tend to stabilise the ion by charge distribution.
As said, the representation of LiMe as an ionic compound is a simplification but one thing is certain: due to the big difference in electronegativity
between Li and C, the Li-C bond is highly polarised, with a strong partial charge residing on the C-atom. This makes LiMe a useful reagent in OC.
Organo-metal compounds like LiMe are particularly useful as reagents towards compounds containing a partly positively charged C-atom, like the C=O
carbonyl group. This double bond is highly polarised due to the difference in electronegativity between C and O.
Here’s an example of the reaction between LiMe and acetone (carried out in an ether-type solvent):
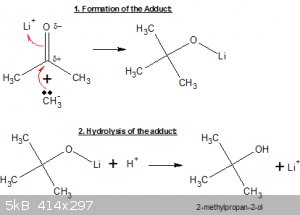
In the first step the lone electron pair of the carbanion latches on to the partly positively charged carbonyl C-atom, while the double carbonyl bond
opens up to the Li cation. An adduct has been formed.
To complete the synthesis, in the second step some weak acid solution is added, which hydrolyses the adduct to a tertiary alcohol.
Note that in the reaction two carbon atoms have been linked together, it’s a so-called C-C coupling reaction.
LiMe has many drawbacks though: its incompatible with oxygen, CO2 and reacts violently with water and alcohols to boot.
Safer, tamer organo-metal reagents have therefore been developed.
Next instalment: Grignard organo-metal reagents.
[Edited on 7-3-2016 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Hello ? Anybody here ? hello ? ello ?
An echo. Good. I must be the first here.
Best make some nitrogen triodide and paint it on certain things ...
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Hmmm... for some time one of the absentees here was you. 
Normal service to be resumed very shortly now.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Dear B & D professors,
OC wise, are those two Cl on 1,4 dichlorobenze not rip-offable, e.g. with conc NaOH, perhaps with a little electrolysis stimulation ?
From what little i vaguely recall, the delocalised electron nature of the benzene ring should make those Cl more vulnerable than if they were bonded
otherwise.
Give them some Na to party with, and well, who knows ? (not me).
That compound came up as a napthalene substitute in mothballs while i was looking for an OTC precursor to Anthracene, which i would still like to play
with seeing as it's an OC semi-conductor.
[Edited on 22-4-2016 by aga]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Welcome back to class. Here is your lesson on aryl halides for the day (using mono-chlorobenzene).
| Quote: Originally posted by aga | | OC wise, are those two Cl on 1,4 dichlorobenze not rip-offable, e.g. with conc NaOH, perhaps with a little electrolysis stimulation?
|
Without a strong electron-withdrawing group (EWG) ortho or para to the halogen, the only way you're going to get a chloro group off of a benzene ring
with NaOH is by reacting them under extremely harsh conditions (like 300 °C at 200 bar/3,000 psi). Cathodic reduction in an electrochemical cell will
remove a halogen from a benzene ring, though, albeit through an entirely different mechanism.
As far as the reaction between aryl chlorides and NaOH goes, if an EWG like -NO2 is present, either in the ortho- or para-position (or
both), NaOH will remove the chlorine by first adding to the benzene ring and breaking aromaticity. The unstable intermediate then collapses back into
a much more stable aromatic system by eliminating either the hydroxyl group or the chloro group. If the hydroxyl group gets eliminated, a nucleophilic
attack by hydroxide can occur again. If the chloro group gets eliminated, the reaction stops. This kind of reaction is a type of nucleophilic aromatic
substitution, and proceeds via the usual SNAr mechanism:
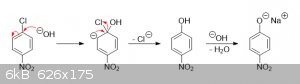
While not shown in the simplified mechanism above, the intermediate of the second step is stabilized by the nitro group like this:
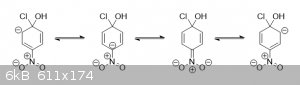
Notice that not only does the nitro group help to further delocalize the negative charge by spreading it out across more atoms, but the nitrogen also
has a positive charge on it, increasing the stability of the adjacent negative charge on the carbon that the nitro group is connected to. And because
the nitro group is strongly electron-withdrawing, it also significantly increases the electropositivity of the carbon opposite of it as well, making
the chloro group carbon much more susceptible to a nucleophilic attack by hydroxide in the initial step of the substitution.
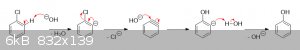
Also, as mentioned above, at high enough temperatures and pressures it's possible for substitution by NaOH to take place even without an EWG; however,
substitution in this manner proceeds through a different mechanism that involves the formation of an extremely reactive benzyne intermediate:
| Quote: | | From what little i vaguely recall, the delocalised electron nature of the benzene ring should make those Cl more vulnerable than if they were bonded
otherwise. |
It's actually the other way around. Because the benzene ring's carbons are sp2-hybridized and the C-Cl bond has partial double bond
character (due to the partial delocalization of one of chlorine's lone pairs into the aromatic ring), the bond length between chlorine and carbon in
chlorobenzene is actually shorter than in alkyl chlorides, making it much stronger and harder to break.
Look at the resonance structures for chlorobenzene:
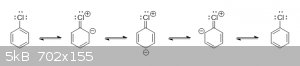
Notice that three of the structures have a double bond between chlorine and carbon. This means that, in reality, the C-Cl bond isn't a true single
bond, and the actual structure of chlorobenzene has a partial double bond between chlorine and carbon.
| Quote: | Give them some Na to party with, and well, who knows ? (not me).
|
Depending on the solvent and conditions employed, sodium metal can indeed remove aryl chloro groups. What happens is that the sodium transfers an
electron to the substrate to give a radical anion, which quickly decomposes into a chloride anion and a phenyl radical. A second sodium atom then
transfers an electron to the phenyl radical to give phenylsodium (an organometallic compound):
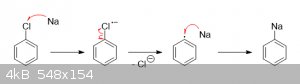
Subsequent reaction of this superbase with a proton source will replace the sodium with a hydrogen.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
So OC is just as hard as Calculus then. OK.
Some bits to clearup if you would be so kind : 'strong EWG' is still a bit fuzzy.
Is that relatively strong, or strong enough to make All the difference reaction-behaviour-wise ?
For the final mechanism you posted, is this more-or-less the same for dry-distilling any benzene-based compound with NaOH to get benzene ?
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Based on this particular chapter I'd say it was the other way around!
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Yes, you're right. Sorry. I'll reverse it:
.KO .neht suluclaC sa drah sa tsuj si CO oS
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by aga | | Is that relatively strong, or strong enough to make All the difference reaction-behaviour-wise ? |
The stronger the electron-withdrawing group is, the easier it will be to substitute the halogen and the less harsh the reaction conditions will need
to be. Having more EWGs on the ring ortho and para to the halogen will also make it easier. For example, substituting chlorobenzene with NaOH requires
reaction temperatures of 300+ °C and pressures exceeding several thousand psi, while substituting 2,4,6-trinitrochlorobenzene can be done under
normal atmospheric pressure at nearly room temperature.
| Quote: | | For the final mechanism you posted, is this more-or-less the same for dry-distilling any benzene-based compound with NaOH to get benzene ?
|
I think there may have been a misunderstanding on that last part. By "give them some Na to party with," I thought you meant elemental sodium. But
looking back, I'm guessing you really meant sodium ions. That last reaction I posted is between chlorobenzene and actual sodium metal. The sodium
metal reduces off the R-X halide group to give an organosodium compound, which is an extremely strong base that could then be protonated to give the
desired R-H group. Blogfast did a lesson on organometallics just a few posts up.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Yes, my language was vague because i really do not know what i'm talking about.
Apologies.
I must have missed that reaction mechanism lesson and will go back and read it.
Thanks for the pointer, and we'll call it quits on the Epcot Centre Incident.
All photos erased.
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
How can we be sure that you won't try to recover them for use in future blackmail attempts? Surely you're aware that nothing's ever truly deleted
simply by emptying the recycle bin. So just to be safe, we are sending someone from IT over to your dorm to scrub your hard drive. A three-pass wipe
ought to be enough.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Three-pass wipes are strictly reserved for the day after the Michlemas Dinner, as Sir should be aware.
Edit:
What happened to the alkycarboxywotsits and all that Electron pushing ?
More Edit:
Strange, but earlier today i went back and re-read a lot of this thread and copied out those group/name structures you posted many moons ago.
[Edited on 1-5-2016 by aga]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
Whenever you're ready for more alkycarboxywotsits, red squiggly lines, equilibrium arrow thingies, snakes and ladder diagrams, nomenclature
mumbo-jumbo etc, just say the word.
Speaking of which, I've been wondering if perhaps these lessons would be more effective if you were the one deciding the curriculum. I was thinking
that maybe you could pick some topics that you think you might be interested in learning more about, either simply out of curiosity or for actual
practical application, or even just as a refresher on stuff we've already covered. We could always go back over the material you're still struggling
with.
Anyway, where we go next is up to you. There's definitely no shortage of OC topics to choose from. If you're still having trouble predicting the
products of some of the more common types of reactions or proposing plausible mechanisms for them along with proper arrow pushing, we could always go
back to some of the reactions we've covered in the past.
We could even go over the OC exam that you took back in December. Unfortunately I was so busy with school at the time that I never got the chance to
really go over the questions you missed like I had intended to. You did appear to have a pretty good grasp on electrophiles and nucleophiles from what
I could tell, which are the very foundation of organic chemistry and the majority of its reactions; however, like gdflp pointed out back in December,
you also seemed to be having trouble drawing the actual mechanisms through which they react with one another, particularly the electron pushing parts.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
gdflp is right !
Yes, it would be great if we could go over the mechanisms again, especially the ones i got totally wrong.
Edit:
It would be awesome if we could do the Theory behind a particular reaction, then do the Practical.
There's still the TCA synthesis so we can convert pinene to turpineol, although that may be a series of difficult cases.
[Edited on 2-5-2016 by aga]
|
|
|
Darkstar
Hazard to Others
Posts: 279
Registered: 23-11-2014
Member Is Offline
Mood: Sleepy
|
|
| Quote: Originally posted by aga | | It would be awesome if we could do the Theory behind a particular reaction, then do the Practical. |
I think blogfast has a lesson on Grignard reagents already typed up that he's been waiting to post. You want to continue on with the organometallic
theme and try a Grignard reaction? They aren't too terribly complicated or anything, but everything must be as dry as possible. A small amount of
iodine to kick off the reaction between the magnesium metal and the alkyl halide will help speed things up as well.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Cool ! Let's go !
My new fume hood is about a week from completion tho ...
|
|
|
| Pages:
1
..
28
29
30
31
32
33 |








