| Pages:
1
..
28
29
30
31
32
33 |
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Again, for anyone experimenting with the salts of DDNR, be extremely careful handling these compounds
Cited from US6946042B2 (Josef kohler):
"In the context of using diazinates, which are known from German Patent 391427 and German Patent 2806599, it must be mentioned for reasons of
safety that the potassium salt of the dinitrodihydroxydiazobenzene in particular is not safe to handle and, similarly to lead azide, tends to
self-detonate during production. This behavior is particularly observed during mixing of water-soluble potassium salts with a diazinate. Strontium
diazinate also shows very high sensitivity to impact and friction in its pure form and therefore may also be poured and processed only with suitable
passivators, in this case strontium sulfate. For reasons of industrial availability of the starting materials and simpler synthetic pathways,
therefore, the non-toxic metal cations of 4-diazo-2,6-dinitroresorcinol are preferred."
The extreme friction sensitivity is is apparent from the table in US4246052 "SnCl2 reduction of Styphnic Acid and DDNP analogue therefrom"
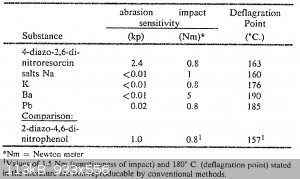
What seems strange is that 4-DDNR is taken as example, while the reduction/diazotization described in this patent is very likely to produce 2-DDNR.
The deflageration temperatures seem odd as well. It is likely that quantity, purity and heating rate of the sample will have a large influence on the
apparent explosion temperature, though, there is a big gap between the 176 deg C from the patent and the 260 deg C, from the potassium salt I got.
It has been described before that the potassium salt precipitates as light yellow needles from concentrated solutions and brown triclinic blocks from
diluted ones. When care is taken to only slightly heat the water solution (60 deg C or so) the potassium salt seems to crystallize as the light yellow
needles irrespective of dilution. From boiling water the brown-blocks are formed again. I think the dinitropyrogallol decomposition product is maybe
acting as crystal modifier, or maybe even form some double salt. It might be interesting to look at possible double salts and complexes for these
compounds, not sure how stable these salts are during storage however. Noticed quite some patents mention the DDNR salts for use in lead free bullet
primers, does anyone know if they are really used in practice? Could find some examples for SINTOX, though not for DDNR salts...
[Edited on 14-1-2019 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Last bit of isopicramic needed to get rid of...
Set out to make made some very pure p-DDNP (4-diazo 2,6-dinitrophenol) using oxidative purification (dilute nitric), after which it was thoroughly
washed with dH2O, dried and recrystallized from cold acetone by slow evaporation in a beaker. The pure product is only a faint yellow-beige. Funny,
all the diazophenols seem to form thin elongated plates or even hair like needles when recrystallized from acid, though both o-DDNP and p-DDNP form
these almost cubic like blocks from acetone. Would these be orthorombic or triclinic, anyone any idea?  (I have no idea how you could see the difference between these crystal forms) (I have no idea how you could see the difference between these crystal forms)
 
Attachment: 4-diazo 2,6-dinitrophenol purified recrystallized.avi (980kB)
This file has been downloaded 912 times
[Edited on 15-1-2019 by nitro-genes]
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Are you going to try for the amorphous, bright yellow form, NG?
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Almost everything I made over the last couple of years was yellow of some sorts, which compound are you referring to exactly?
|
|
|
hissingnoise
International Hazard
Posts: 3940
Registered: 26-12-2002
Member Is Offline
Mood: Pulverulescent!
|
|
Indeed, the nitrated benzene derivatives are a colourful lot ─ and Davis in COPAE notes that when DDNP in hot acetone, is precipitated by
adding a large volume of ice-water to the rapidly agitated liquid it is an amorphous, bright yellow powder...
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
It depends on the isomer, o-DDNP often precipitates as more of a bright yellow colour, while there is a definite beige tint for the p-DDNP. In this
case though, I just wanted to make some really large crystals (just for the fun of it). Precipitation from acetone ice/water also works well, though
cleaning first and then slow evaporation was obviously much easier in this case. Was curious if a really large crystal would be able to make DDT on
its own. The diazotization of both picramic acids always seems to produce some brownish impurities, that can be removed by warming in nitric or
water/nitrosylsulfuric. During the evaporation from acetone, these impurities could precipitate along with the p-DDNP , so to make really large and
pure p-DDNP crystals these had to be removed. Was also curious if any impurities would arise from reaction with the acetone, especially in the
presence of traces of acids from diazotization, though no colour change during the evaporation (which took 3 days at 10 deg C in the dark!) was
observed, the acetone solution remained the same light yellow colour.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Quote: Originally posted by nitro-genes  | Styphnamic acid!!!  : :
The copper/ascorbic method of reducing picric acid to picramic acid seemed surprisingly specific in reducing one ortho nitro group (the method was
posted on page 28 of this thread), even approaching the efficiency of the well known (hydro)sulfide reduction. I was curious whether the
copper/ascorbic reduction would produce similar results for styphnic as for picric, as also mentioned by Rosco Bodine a while back. The product upon
reduction of styphnic would be styphnamic acid, a pretty elusive compound, which is, (similar to it's diazo derivatives) only few times mentioned in
literature. The diazo derivatives itself are of little interest as energetic materials in practice due to incompatibility/stability issues and the
reported extreme friction sensitivities of their salts. The chemistry of both these selective reductions and diazo derivatives is still very
interesting IMO.
Basically the same approach for the reduction of styphnic was used as that for picric posted earlier. The styphnic was produced by hydrolysis of
3-aminopicric using 3 molar eqvts of NaOH and boiling the solution until no more smell of ammonia could be noticed (~30 min) followed by acidification
using HCl. Overall, the reduction of styphnic seems to behave very similar as that of picric. Curiously, whereas the reduction of picric produced
very dark brown solutions and produced a lot of gas during the reaction, the reduction of styphnic was without any noticable gas formation and no dark
brown solution was observed near the end of the reduction. Yield needs to be measured, though might be at least as good or even better as that for
picric.
Experimental:
Reduction of styphnic to styphnamic acid:
0.5 g of styphnic was added to a 100 ml beaker and water was added to 75 ml in total. Then, 0.27 g basic copper carbonate was added and the solution
heated until all styphnic had dissolved into a dark yellow solution and no more CO2 was produced. A bit of undissolved copper carbonate remained
because of the slight excess used. The copper styphnate solution was cooled to about 10 deg C. Next, 1.5 g ascorbic acid was weighed out and added at
once. After the ascorbic had dissolved, I let the reaction stirr in the cold for another 10 minutes, though no clouding or precipitation was observed.
The hotplate was turned on and the solution was heated slowly. When at 25 deg C. Slight clouding could be seen forming first, followed quickly by a
light green precipitate (very similar to that observed for the picrate reduction). Heating was continued and slowly brought up to about 65 deg. C. The
light green precipitate started to colour increasingly more yellow-greenish-brownish (also similar to the picrate reaction). After 30 minutes at 60
deg. C., the mixture was cooled down to room temperature and the precipitate left to settle. Most of the supernatant was decanted, then another 75 ml
of cold water was added and most decanted/siphoned off again. The beaker was added to the hotplate again, 10 ml water was added and warmed slightly
while dropwise additions of concentrated HCl were started. The greenish-yellow precipitate slowly turned a more orange colour and dark copper-reddish
crystals of styphnamic acid started forming. The solution was cooled down in the fridge, filtered and washed with water to remove all of the copper
and finally dried. The crystals of styphnamic acid look very similar to picramic, though with a more golden-brown note to it (See attached photo, some
cuprous styphnate seems to still be present). A rough melting point was taken on the hotplate and had a much higher melting point as picramic acid at
around 210-220 deg. C. (with extensive bubbling and decomposition).
Diazotization to dinitro diazoresorcinol or DDNR:
About 100-200 mg of the stypnamic acid was added to a 10 ml beaker together with 5 ml of 10% HCl. Some of the styphnamic acid dissolved, most remained
as suspension. This was cooled to 0 deg C. in an icebath and 2 ml's of water with a spatule of sodium nitrite dissolved was added dropwise. Here is
where it gets weird... Each drop of the nitrite solution produced a pretty dark
colour (maybe due to residual copper?, or an N-nitroso intermediate?). After only a few drops of the nitrite solution were added, all of the
styphnamic acid dissolved into a clear dark yellow-brownish solution. After stirring for another couple of minutes, a very light yellow precipitate
started to from (chloride salt of DDNR?). Thinking it was the DDNR itself, I reasoned adding another 5 ml of cold water would probably precipitate
more of the DDNR. To my surprise however all product dissolved again. Since there hadn't been any gas production during the diazotization itself, it
seemed unlikely the compound had decomposed somehow, as could have been the case for styphnamic containing an amino group in 2-position. To
precipitate any possible DDNR as the salt, solid sodium bicarbonate was added in small increments. When most of the HCl was neutralized, large amounts
of fine dark yellow needles started to precipitate, presumably the sodium salt of some DDNR isomer.
Since these salts are extremely dangerous (and I had made quite too much  ) I
saved a few mg's and dissolved the rest in warm water again. When dry it behaves very energetic though, detonating in tiny amounts, much resembling
SADS. ) I
saved a few mg's and dissolved the rest in warm water again. When dry it behaves very energetic though, detonating in tiny amounts, much resembling
SADS.
What really puzzles me is how the presumed internal diazonium salt of styphnamic acid (DDNR) can actually seem more soluble in cold HCl as the
styphnamic acid itself...Also curious which isomer is formed during the reduction, maybe that could explain things....hmmm....It also doesn't seem
like the same compound as was obtained from the nitration of iso-picramic acid....pfff, I was expecting to close the loop and connect all dots here,
and then this?!?!?! ...if anyone has some thoughts on this, please share
them!!!
[Edited on 25-11-2018 by nitro-genes] |
An idea I had is wondering if formaldehyde may react with the DDNR in similar fashion to form a compound or polymer as "resorcinol glue", and this may
be a way to desensitize the DDNR.
Also had an idea that hydrazine, or methylamine, or trimethylamine or tetramethylamime, or aminoguanidine, di or tri variants could be used as
desensitizers, looking at organic amine salts as opposed to metallic salts could be an approach that may be useful.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
My guess would be that due to the nitro groups, the ring system is too electron deficient for formaldehyde to attack any of the aromatic carbons
directly. My guess would be that the carbon of the formaldehyde would attack the outer nitrogen of the diazogroup bearing a slight negative charge.
Then several things could happen (perhaps depending on pH and solvent used), electron transfer from the nitrogens of the diazogroup to the ring,
forming a transient imine which would maybe hydrolyse leaving some hydrazone and dinitropyrogallol. Alternatively, it would lead to deamination of the
ring by electron moving from the diazo group to the aldehyde, producing N2, formic acid and leaving 4,6-dinitroresorcinol as the product. The latter
would be interesting as a route to the 4-diazo derivative of DDNR, as I am pretty sure I've seen at least one patent reagrding the resonably selective
nitration of resorcinol diaacetate to the 4,6 dinitro derivative and mono reduction using hydrazine.
[Edited on 31-1-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Might be worth checking reactivity of the soluble calcium DDNR with urea to see if a low soluble urea DDNR salt may exist. Bivalent / trivalent metals
possibly could form complex "designer" salts .... Al (DDNR) styphnate or Pb
(DDNR) picrate or basic salts or metal complexed salts might be another scheme to reduce sensitivity for a DDNR component in the compound.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The lithium and rubidium salts might be interesting as well, very probable these were made in some lab, some time before though, probably
hydrates/impossible to dry, hygroscopic or similarly sensitive as the other alkali metal salts. There could be some interesting complexes possible
with the DDNR salts, they are to sensitive for my taste though. It is possible that the reported extreme sensitivity for the salts of 2-DDNR is in
part due to the very fine needle like crystal morphology (similar to lead azide) of the K-salt when formed from water and a potassium salt, though
they probably remain pretty sensitive regardless. Even when diluted 50% with the sulfate as Dave321 and one of these patents mentions IIRC, the
strontium salt is still considered a very friction sensitive composition.
[Edited on 31-1-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The ferric salt and double salt with styphnate or picrate would be an obvious candidate, maybe first make the basic ferric styphnate or picrate and
then try converting to the neutral compound salt using free DDNR. Similar scheme could be applied to other metals maybe zinc and manganese for
example.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Not sure if any of the transitions metals would make suitable salts, I wonder if these would be very stable, some would probably hydrolyze (Fe, Al, Ti
etc), others may not, though may be facilitating reactions of the diazo group with coordinated water or OH- resulting in less stability maybe? Maybe
there just too soluble or something, no idea why they are not mentioned in literature exactly...
The potassium salt of 2-diazo 4,6-dinitroresorcinol is described to crystallize as fine yellow needles, though when recrystallized from boiling water
as brown triclinic blocks. IIRC, the article describing this phenomenon suggest a hydrate of the potassium salt is formed. Maybe interesting to see
what the brown blocks from boiling water are exactly, would be interesting if this represents a double salt of the potassium salt of
4,6-dinitropyrogallol and the potassium salt of 2-diazo 4,6-dinitroresorcinol. It could be that the observed decrease in the mechanical sensitivity of
the brown compound (compared to the light yellow potassium salt of 2-DDNR) and the elemental analysis together were incorrectly interpreted as being a
hydrate of the potassium salt. Styphnates form all sorts of double salts as well, maybe stypnic, or 4,6 dinitropyrogallol double salts for calcium,
potassium, barium, strontium etc, maybe nickel or cobalt might be interesting indeed. Though how to determine if you've made a true double salt or
just mixture? Any free salt of 2-DDNR would render the mix much more dangerous to experiment with probably. Sort of a clathrate compound, similar to
leadpicrate/leadzide ones also might be possible.
[Edited on 3-2-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
The half neutralization of ethylenediamine to form a monoperchlorate with the further neutralization by picric acid, illustrates a reaction scheme
that may be much more general and could be applicable to use of DDNR as an included component in a complex energetic compound which would serve to
tame the sensitivity of the DDNR, yet retain its energy.
http://www.sciencemadness.org/talk/viewthread.php?tid=13174&...
For example if the scheme using ethylenediamine was adapted then a compound salt could result that is ethylenediamine DDNR perchlorate, or other
substitutents than perchlorate may be workable.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Somewhat offtopic:
It seems the amino group of styphnamic acid (2-amino 4,6-dinitroresorcinol) is more suceptible to hydrolysis than I thought... While boiling of
styphnamic with sodium carbonate only results in dark brown black resin formation, prolonged boiling of styphnamic acid in dilute acid seems to result
in hydrolysis to 4,6-dinitropyrogallol (4,6-dinitro 1,2,3-trihydroxybenzene). Is picramic acid equally sensitive to acid hydrolysis?
Does make for a nice synthesis of 4,6-dinitropyrogallol though:
Experimental:
0.5 g of 2-amino 4,6-dinitroresorcinol was added to 80 ml distilled water and 1 ml 97% sulfuric was added. The suspension was boiled for 3 hours while
covered with cling wrap upon which a light yellow solution had formed. Upon slow cooling, 1 cm long bright yellow crystals formed (attachment 1), that
turned a more orange shade upon washing with water. It melts sharply at about 210 deg C (without visible decomposition), with immediate sublimation
after melting. Presumably 4,6-dinitropyrogallol in nearly quantitative yield. Maybe some interesting salts or pyrotechnic mixtures could be made with
this stuff. It does not behave very energetic, only burns slowly with a luminous flame when strongly heated (near it's melting point).

Also seems 4,6-dinitropyrogallol forms some interesting product when reacted with nitrous acid in concentrated HCl:
Experimental:
100 mg 4,6-dinitropyrogallol was added to a 10 ml beaker and suspended in ~1.5 ml 20.2% HCl. The beaker was added to an icebath and while stirring, an
excess of sodium nitrite in water was added dropwise. Some of the 4,6-dinitropyrogallol dissolved and a pretty bright orange compound was formed
instead (Attachment 2). I puffs off pretty violent when heated to ignition. The orange compound formed seems very unstable, already at ~50 deg C, it
starts to blacken very fast, though it remains pretty explosive. It seems to form a tarry substance, as black as coal, when at 100 deg C, though it
still remains pretty explosive. Finally it melts at around 120-130 C or so. maybe some dinitro dihydroxy quinone? An isomer of nitranilic acid maybe
(that would be cool)? Some chloro derivative? The product seems pretty pure and crystalline, maybe just a mixture of something with unreacted
4,6-dinitropyrogallol? Any ideas?

[Edited on 8-2-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Maybe you got a 5-nitroso addition product ? Try nitrating the putative 5-nitroso using plain concentrated HNO3 and perhaps very gentle heating and
the nitroso may convert to the nitro giving the trintropyrogallol. This would be isomeric with trinitrophloroglucinol.
If that proves true then it could be interesting to do a second ascorbic reduction, and then diazotize to obtain a "hydroxy" DDNR
which would be a dibasic acid and should form energetic salts.
This potential compound may be a novel and unreported isomer of the phloroglucinol analogue reported in 1923 by Von Herz in GB207563
[Edited on 2/10/2019 by Rosco Bodine]
|
|
|
MineMan
International Hazard
Posts: 1004
Registered: 29-3-2015
Member Is Offline
Mood: No Mood
|
|
I am kind if lost, are we talking about different routes to prepare DDNP... or it’s novel cousins? Would ligands be the right word??
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
We are talking about a specific cousin and analogue / variant / hydroxylated form of DDNP that is more powerful.
DDNR and isomers are Hydroxy substituted variants of DDNP, and are acidic phenolic compounds able to form salts because of the free phenolic hydroxyl
being present, while the diazo-oxide bridge has tied up the one other hydroxyl making DDNP neutral. DDNR has one free phenolic hydroxyl and is a
monobasic acid like picric acid.
With a trinitrotrihydroxybenze like derived from phloroglucinol or pyrogallol, having one nitro reduced and the resulting dinitroaminotrihydoxybenzene
diazotized, what then results is a compound like DDNP but having 2 free phenolic hydroxyls, providing a more energetic form of DDNP that is a dibasic
acid for the 2 phenolic hydrogens available, and able to form salts.
I was speculating that the orange compound nitro-genes shows above is an oxime or nitroso precursor that will easily nitrate to trinitropyrogallol on
gentle warming with medium concentrated HNO3, which may then be reduced to the monoamino derivative by ascorbic acid and copper, and that same
monoaminodinitrotrihydroxybenzene could then be diazotized to provide the "hydroxy-DDNR" dibasic energetic acid. This would be an isomer of the
phloroglucinol derivative already known and identified by Von Herz in 1923. The compound I speculate could form would be the pyrogallol analogue for
the Von Herz phloroglucinol derivative which is evidently unreported and novel, but seems workable by this proposed reaction scheme.
If the novel compound exists, it should go into the journals. It may be that the speculated pyrogallol derivative compound is already reported long
ago under some ancient synonym since the old nomenclature gets bizarre and confusing.
[Edited on 2/11/2019 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Not sure how readily and stable nitro-nitroso substitution of the 5 position for 4,6-dinitropyrogallol would be. At first glance, both the 1,3 hydroxy
and 4,6 dinitro groups seem to work against any presence of a nitroso/nitro. It seems likely that any transient nitroso addition compound would
immediately be hydrolysed to leave the quinone that would be an isomer of nitranilic. IIRC, only the trinitro derivative of the trimethylether of
pyrogallol has been described, though hardly any properties were given. On the other hand, benzoxazolones actualy favour the electrophilic substition
of the 5,6 positions ....similarly, 5,6-benzofuroxan is more stable as the 4,6-isomer IIRC. Benzene chemistry is really strange... One thing that I do seem to remember reading in some book on quinone toxicity is that
the isomer of nitranilic acid was insanely toxic, this is from a vague memory years ago and I cant find it again. Is there a good source for toxicity
data of these compounds?
[Edited on 11-2-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
You know you want to put the orange stuff in some concentrated HNO3 to see what happens curiosity / cats 
On toxicity nitroso compounds are a good bet to be toxic and carcinogenic
In my long experience, caveat emptor.... test the unknown sample first to see if it is finger licking good ......you just never know what you might be
getting into there
[Edited on 2/11/2019 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Indeed, but then again...there aren't many things I wouldn't want to add to concentrated nitric.
The 2-diazo 4,6-dinitroresorcinol behaves very different in many aspects as the "others" so to say. Surprisingly, it seems the ammonium salt can
actually be isolated and dried. When a solution of 100 mg of 2-DDNR was added to about 2 ml of water and slowly dripped in 5 ml of a 10% ammonium
acetate at room temperature, everything stayed in solution first and no nitrogen was evolved at all. When cooling on ice for a few minutes, it
precipitated almost quantitatively as long and glossy light yellow needles. It burns only weakly energetic in small amounts, though becomes
increasingly more so with larger amounts, as can be seen from the video. On heating a few mg it just puffs off with white/yellowish flash. The
ammonium salt is very soluble in warm water, and very insoluble at 0 deg C. I was so suprised to see how mildly energetic it was, that I was first
thinking some internal rearrangement had taken place. When the ammonium salt is added to water again, and added to excess of a KCl solution, the
extremely explosive potassium salt forms again. It really is a salt, maybe hydrate, amazing to see how that hydrogen bonding is able to stabilize the
compound. Wonder how stable it would be on storage, one would think the ammonium would eventually react with the diazonium group right? If not, the
ammonium salt or maybe the ethylenediamine salt might be used as a transfer reagent, similar to 5-NT.
If one is to draw the structure of the salts...how would it best be drawn? Does the cation combine with the delocalized negative charge of the ring,
as sort of a meisenheimer complex, or just with one of the 2 hydroxy groups?
Attachment: Ammonium salt 2-Diazo 4,6-dinitroresorcinol - Copy.avi (929kB)
This file has been downloaded 891 times
Starting to wonder how unstable the salts of this thing are exactly, it happily sits at a pH of >12 with hardly any nitrogen gas formation. Wonder
if it is more unstable at acid pH, which could be helpful for destruction. Maybe do a timed boil test in water and check for presence of potassium
salt after fixed time intervals. Maybe sulfites would do better?
[Edited on 13-2-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
You should see how the ammonium DDNR reacts to slight confinement, like aluminum foil, and if it will DDT in some small critical mass.
The ammonium DDNR should be stable and it might be a safe storage form for the DDNR to be kept as a precursor for other DDNR salts.
Since the ammonium DDNR appears benign it should be tested what would be the Cupriammonium DDNR by reacting the soluble Ammonium DDNR with copper
nitrate, and the nickel salt and cobalt. Silver is another possibility.
The possibility of a double salt of the potassium DDNR with another energetic salt is still intriguing, since it would likely be a synergy that would
retain the energy of the potassium DDNR while possibly reducing the sensitivity. Possibly a mixed equimolar solution of calcium DDNR and calcium
picrate being added into solution of KNO3 could form a bridged compound double salt of potassium DDNR and potassium picrate leaving byproduct calcium
nitrate in solution. If it works to form a double salt the compound could have favorable properties.
[Edited on 2/13/2019 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by nitro-genes |
If one is to draw the structure of the salts...how would it best be drawn? Does the cation combine with the delocalized negative charge of the ring,
as sort of a meisenheimer complex, or just with one of the 2 hydroxy groups?
|
The diazo-oxy bridge is a dehydrogenated hydroxyl group so there is only one intact hydroxyl on DDNR and it becomes dehydrogenated also when a salt
formation occurs and a metal or other cation replaces the phenolic hydrogen there.

Really while it is interesting to see what further can be done with the o-DDNR isomer that is more commonly known .....I still think the more obscure
isomer p-DDNR derived from nitration of paracetamol acetate followed by hydrolysis and diazotization may gain enough stability by virtue of the 4-1
positions diazo-oxide bridge that the potassium salt will be less sensitive and more useful than the overly sensitive potassium o-DDNR. Acetic
anhydride is the missing ingredient.
Two years ago I believe I had sorted this out concerning the structure.
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
https://www.youtube.com/watch?v=q-0bQKoXD2k Do it Again
[Edited on 2/14/2019 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
The electronic structure of diazophenols have been reviewed a couple of times. None of them mention a cyclic structure, although in this case it
surely seems convenient indeed.
https://www.youtube.com/watch?v=hPvLYLQoN0A
Maybe only the para diazophenols behave more as a zwitter ion (more charge separation means more energy?) and the ortho more as quinones or cyclic,
just don't know enough of physical chemistry to determine how convincing the data in all of them are as proof of their structure, it is an interesting
topic for sure. The article from 1987 is the most modern review on this topic AFAIK, it mentions complete delocalization of the negative charge over
the entire ring, which would be more in line with also the electronic structure of nitranilic acids and other salts of dihydroxyquinone IIRC. Still
having trouble to see where that metal cation will end up exactly Other sources
mention partial charges that change with the nature of the substituents and pH, I have really no idea where all these partial charges would end up to
form the salts of the DDNRs though.
Kazitsyna, L. A., B. S. Kikot', and A. V. Upadysheva. "Quinone diazides and p-iminoquinone diazides." Russian Chemical Reviews 35.5 (1966): 388-405.
Kazitsyna, L. A., and N. D. Klyueva. "Electronic structure of substituted diazophenols." Russian Chemical Bulletin 19.1 (1970): 197-199.
Lowe-Ma, Charlotte K., Robin A. Nissan, and William S. Wilson. Diazophenols-their structure and explosive properties. No. NWC-TP-6810. NAVAL WEAPONS
CENTER CHINA LAKE CA, 1987.
An other interesting reference in relation to the cuprous mediated reduction seems the one below:
Porter, Thomas R., et al. "Sterically directed nitronate complexes of 2, 6-di-tert-butyl-4-nitrophenoxide with Cu (II) and Zn (II) and their H-atom
transfer reactivity." Dalton Transactions 46.8 (2017): 2551-2558.
Cuprous ions can maybe form some transient SET complex with the quinone-nitronate resonance form of styphnic to facilitate the reduction. Cuprous can
form very strong ligand interactions (almost like covalent) so this explains maybe both the efficiency of the cuprous reduction as well as why the
2-nitro is selectively reduced? It would maybe also explain why styphnic acid seems more readily reduced at lower temperatures as picric using the
cuprous method?
[Edited on 14-2-2019 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
How sure are you it even actually *is* the 2 nitro being reduced?
| Quote: Originally posted by nitro-genes |
The extreme friction sensitivity is is apparent from the table in US4246052 "SnCl2 reduction of Styphnic Acid and DDNP analogue therefrom"
What seems strange is that 4-DDNR is taken as example, while the reduction/diazotization described in this patent is very likely to produce 2-DDNR.
[Edited on 14-1-2019 by nitro-genes] |
Where *exactly* do you get this stuff about *2*???- DDNR ????
Attachment: Diazophenols-their structure and explosive properties. NAVAL WEAPONS CENTER CHINA LAKE CA, 1987.pdf (1.3MB)
This file has been downloaded 728 times
[Edited on 2/14/2019 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Not 100% sure, though I'm not 100% sure the earth isn't flat either . The
melting point of styphnamic is described as 190 C by Benedikt and Hubl, though as 219 C by the chinese article from 1933 (abstract link attached),
which is alot more modern. The abstract (see link below) also mentions boiling in water produces dinitropyrogallol as proof of structure, something
Benedikt and Hubl didn't provide. The melting point of the styphnamic from the copper/ascorbic reduction seems around 220 C as well, dilute acid
hydrolysis produces quantitative yield of a yellow compound melting at around 210 (commercial mp. 4,6-dinitropyrogallol = 209-211C), the left over
filtrate when all of the presumed dinitropyrogallol is removed produces an ammonia smell with NaOH, so likely an amino hydrolysed off. It is very
unlikely that p-amino phenol derived diazo derivatives somehow form 2-amino resorcinol derivatives, and the 4-DDNR product from these seems much less
soluble as the presumed 2-DDNR, as it precipitates from very dilute acid solutions (Klapotke), while the presumed 2-DDNR has an incredibly high
solubility in water. Benedict and Hubl also described the diazo derivative from their reduction product to be only precipitated from concentrate
solutions and concentrated acids, which also fits the observations of the compound I obtained. Combined with the likely preference for ortho
hydroxy-nitro reduction described by Porter and the presumed quinone-nitronate structure mediated reduction, which fits the huge preference for the
2-nitro, this is the absolute most likely scenario, so without any constructive arguments I'll stick to the 2-DDNR. The only remote possibilities
seems that the stannous chloride reduction of styphnic produces somehow the 4-amino reduction product and that the 2-amino of styphnamic may
rearrange to take the position of one of the other hydroxy groups, AFAIK, this has never been described though for similar compounds.
[Edited on 14-2-2019 by nitro-genes]

|
|
|
| Pages:
1
..
28
29
30
31
32
33 |








