| Pages:
1
..
28
29
30
31 |
B(a)P
International Hazard

Posts: 1139
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
Quote: Originally posted by ManyInterests  |
I took the remaining water (around 350ml) and I will evaporate it down until it is around 150ml or so, filter off whatever crystals I find, then
discard the remaining liquid.
|
I would suggest discarding the whole 350 ml and everything it contains given the amount of acid you had. There will not be significant quantities of
picric acid in it.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
Yep. There isn't. I evaporated it down to slightly under 200ml and it has been in the fridge for several hours and is near 0C. Nothing is coming out.
I am going to dilute it, neutralize it with baking soda, then pour the whole thing down the drain.
I am quite proud of cleaning this up and getting consistent crystal shapes. I will be doing a recrystalization of the other batch I had lying around
and slowly removing the sulfuric acid with lots of paper towel changes (so it is mostly acid free, but not quite. I will also need to recrystalize it
to the 400ml level and then discard the rest, as THAT water is bound to have some H2SO4 left in it).
Then after that it'll be making picric acid using my potassium nitrate supply (since I am currently out of sodium nitrate and I am having some issues
with synthesis) and using the salicylic acid that I bought off amazon instead of ASA (which I have no more of and I am not in the mood to through the
expensive and laborious route of getting it from aspirin pills at this time).
I will be using 2.8 grams of potassium nitrate and 4ml of H2SO4 to 1 gram of salicylic acid. An excess for sure, but I want results. It wouldn't be
the first time I made picric acid with potassium nitrate.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
OK so I recrystalized the final batch I did last December (before my 2 liter beaker broke) and I realized that sulfuric acid can be 'cleared' in a way
with changing paper towels frequently. Granted that is a very slow and laborious way of doing things so I won't be repeating it. The paper was mostly
clear once I dumped the picric acid into my new 2 liter beaker for recrystalization. It was made from 50 starting ASA, but I poured in 1800ml of water
and boiled it down to around 475 or 500ml of water before decanting it. I saw the VERY red blob at the bottom that I was careful to leave behind.
I still see some redness in the picric. Is that a problem when I recrystalize or no? I'm legit curious here.
For the remaining liquid, since I am confident that there is going to be some H2SO4 still left in there, I will only boil down the recrystalization
liquid slightly before seeing if there is any viable picric acid crystals I can save. Otherwise it will be discarded. The solution was still very hot
when I took these photos so there is going to be a lot of coming out once it cools.
 
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
So I am back to making picric acid, this time my biggest synthesis. I originally just wanted to do 4 more 75 grams synthesis, but I decided it would
be a waste of time and I could do 150 grams at a time.
I started using salcylic acid and I think it is MUCH better than ASA as I have done previously. I can't describe it, but it seemed to really work much
better than ASA. More nitrates were needed, but that is not an issue I find.
So I had my gas mask on and maybe some of the fumes overwhelmed the filter and I ended up having some minor inhalation, but nowhere near as bad as the
previous times. I fear that my filter is bust. I will be ordering a replacement in the future, but for now I will need to rely on it but also do what
I did previously and maintain as much ventillation as possible, including doing as much of the nitration outside as possible as I did previously (NO2
inhalation was never an issue with my old system).
The main thing is that I learned really hard that the temperature MUST be kept up! While adding nitrates is an exothermic reaction. I noticed that it
is not an exothermic reaction after 50% or so of the nitrate salts are added and the temperature will continue to drop. The result of this is that I
had to put the beaker back on heating to get the temperature up to 95+C again. I also noticed that keeping the heating up does some good in keeping
picric acid's well known foaming issue at bay. It does not eliminate it, but it does keep it more under control. I will need to do this for the future
with my 2nd and final 150 gram synth as I nearly had a foam overflow. Reducing the amount of nitrate addition at a time will also do that. I do not
like overflows!
I am committed on doing it a 2nd it even if the yield I seem to have here is very high. Like I was completely not expecting this kind of stuff. It
must be salicylic acid since the nitrate salts I am using is from the previous batches. The sulfuric acid is obviously just the regular I've been
using.
All in all I am VERY happy with all this. I will make another update next week after filtration and the 2nd batch is done. After that I will dry them
and recrystalize them piecemeal since I cannot recrystalize this much picric acid at one time (my largest container is a 2 liter beaker. So 75 grams
of picric is the most I can recrystalize at any one time).

|
|
|
Herr Haber
International Hazard
Posts: 1236
Registered: 29-1-2016
Member Is Offline
Mood: No Mood
|
|
Do it outside if possible.
NOx filters are not cheap, hard to find and dont last long. Prolong it's life by getting a more generic filter for your "every day use".
I remember you said you used one of Roscoe's synthesis. Apparently a short version (that works of course).
I think you would enjoy doing it in three steps "the slow way" at least once. That's just a personnal vibe I get from reading you. Plus, you'd be able
to control foaming a lot more after that.
About adding nitrate and decomposition: of course you want to add a little at a time. Some of the NOx are unavoidable in this synthesis, but say you
add too much nitrate in one go and you'll lose it as NOx too.
Nitric acid's boiling point is 83c, so if you're heating your nitration mixture at 95 some will get lost not matter what.
Good luck with recrystallization. I wouldnt want to be in your shoes, that's gonna be long 
The spirit of adventure was upon me. Having nitric acid and copper, I had only to learn what the words 'act upon' meant. - Ira Remsen
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: Originally posted by Herr Haber | Do it outside if possible.
NOx filters are not cheap, hard to find and dont last long. Prolong it's life by getting a more generic filter for your "every day use".
I remember you said you used one of Roscoe's synthesis. Apparently a short version (that works of course).
I think you would enjoy doing it in three steps "the slow way" at least once. That's just a personnal vibe I get from reading you. Plus, you'd be able
to control foaming a lot more after that.
About adding nitrate and decomposition: of course you want to add a little at a time. Some of the NOx are unavoidable in this synthesis, but say you
add too much nitrate in one go and you'll lose it as NOx too.
Nitric acid's boiling point is 83c, so if you're heating your nitration mixture at 95 some will get lost not matter what.
Good luck with recrystallization. I wouldnt want to be in your shoes, that's gonna be long
|
Yeah it is going to be long. I just started adding water to the whole thing. I calculated that there is around 1100ml in volume, so I will need 770ml
of water to break everything up. It'll take a while to add everything since I need to do it piece meal and put it back in the fridge to cool down (I
did see some warming after adding 100ml or so). I won't be having any of that flash boiling!
As for doing it outdoors. I want to and I will do it, but it is COLD. So I hope for a day that isn't too cold and I can do the entire thing out side.
But I will also have to consider that I will need to heat it up. I am aware that heat degrades the nitrates, but one thing I am very much afraid of is
that if I add the nitrates when they're cold, I will have a major runaway when I need to heat it up even a little. This actually happened to me once.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
So I did the final picric acid synth and after speaking with someone on a discord group with extensive experience in making picric acid and they told
me to try something differently: instead of adding the nitrate salt while it is hot, I did it while it is cold, and this is the result. I haven't done
the heating step yet, and I won't do it for another hour or so at least to make sure that nothing is going wrong. Albeit so far (around 30 minutes in)
I see that it is going to be OK.
The NO2 was minimal compared to the last time, but it did happen. I was indoors the whole time and there is some NO2 scent in my apartment, but it is
fading fast. I think it is going to be good.
I pray that no runaway will happen during heating. In order to prevent that, I must take absolutely sure that heating is done very, very, VERY
gradually. No more than 10C increments at a time.
Edit: I saw a bit more foaming happening when I was waiting. I realize I need to give it many more hours to make sure all reactions that can happen
have happened before heating. The last thing I want is a runaway with this much picric acid!
  
[Edited on 20-2-2023 by ManyInterests]
|
|
|
Bert
Super Administrator
Posts: 2821
Registered: 12-3-2004
Member Is Offline
Mood: " I think we are all going to die. I think that love is an illusion. We are flawed, my darling".
|
|
| Quote: Originally posted by ManyInterests | So I did the final picric acid synth and after speaking with someone on a discord group with extensive experience in making picric acid and they told
me to try something differently: instead of adding the nitrate salt while it is hot, I did it while it is cold, and this is the result. I haven't done
the heating step yet, and I won't do it for another hour or so at least to make sure that nothing is going wrong. Albeit so far (around 30 minutes in)
I see that it is going to be OK.
The NO2 was minimal compared to the last time, but it did happen. I was indoors the whole time and there is some NO2 scent in my apartment, but it is
fading fast. I think it is going to be good.
I pray that no runaway will happen during heating. In order to prevent that, I must take absolutely sure that heating is done very, very, VERY
gradually. No more than 10C increments at a time.
Edit: I saw a bit more foaming happening when I was waiting. I realize I need to give it many more hours to make sure all reactions that can happen
have happened before heating. The last thing I want is a runaway with this much picric acid!
[Edited on 20-2-2023 by ManyInterests] |
If you are doing this synthesis INDOORS without a hood/air handling, you are risking your health/life and possibly the health and lives of others.
Yes, you're likely to have issues as you heat the mixture. Foaming and/or a runaway are possible. But I'd worry more about your LUNGS.
Note on attached image that the sensory threshold may be as low as .1 ppm, but also note that in cases of slowly raised concentrations ABILITY TO
SENSE LOWER BUT STILL DAMAGING LEVELS IS LOST. The max for a 4 hour exposure may be set at .5ppm, but you can't judge that by your sense of smell.
And it sounds EXACTLY like what you're probably going to be doing, trying to stretch this reaction out and run it without a proper hood and indoors.
Stop. Take it outside. Don't run such nitration reactions indoors unless you've got hood or other effective air handling equipment.
[Edited on 2-20-2023 by Bert]
 
Rapopart’s Rules for critical commentary:
1. Attempt to re-express your target’s position so clearly, vividly and fairly that your target says: “Thanks, I wish I’d thought of putting it
that way.”
2. List any points of agreement (especially if they are not matters of general or widespread agreement).
3. Mention anything you have learned from your target.
4. Only then are you permitted to say so much as a word of rebuttal or criticism.
Anatol Rapoport was a Russian-born American mathematical psychologist (1911-2007).
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
I didn't have that many problems as you are describing, but I am really well aware of the dangers. I will never, EVER do another nitration indoors. No
matter what. My gas mask stopped most of the gasses, but not all, which is why I didn't have much problems this time. But I am extremely well aware of
the dangers. I have had NO2 inhalation issues and I REALLY FUCKING HATE IT!
I spoke with a doctor about it and they did advise more precautions.
I realize that I simply must take seasonal breaks if need be. Winter time is a problem in my area not just due to the cold but often times due to the
snow (electronics and moisture don't mix well). I must accept that there is no shame in waiting a few months before doing something like this.
At any rate, my 2nd synth is fully complete, and the weather outside was well enough that I could do the heating step outside (and it was essential).
I ran into difficulties with my thermometer which was often inaccurate, but I took great precautions to make sure that I was heating everything SLOWLY
and in gradual steps to allow for any reaction to happen piecemeal and prevent a runaway.
When I heated it to 75C I decided to go all the way and when it reached 100C it did start to bubble with red NO2 gasses forming, but I did it outside
and the wind was on my side so all dangerous gasses were blown away.
It has cooled down to near room temperature now and you can see it in the photo. I am not going to do any more picric acid synths for a very long
time. I got too much already and nothing more to prove or explore with it.
I'll be fine, and I will be taking a break from most chemistry for a good long time to focus on other things.
For this and my previous synth, I will make sure everything is dried up before doing a recrystalization of everything and post my final results here
when that happens.

|
|
|
Herr Haber
International Hazard
Posts: 1236
Registered: 29-1-2016
Member Is Offline
Mood: No Mood
|
|
A bit out of topic but anytime I used nitric acid and / or was exposed to NOx I could smell it almost everywhere for a few days.
Buses running on diesel smell specially strong.
The spirit of adventure was upon me. Having nitric acid and copper, I had only to learn what the words 'act upon' meant. - Ira Remsen
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: Originally posted by Herr Haber | A bit out of topic but anytime I used nitric acid and / or was exposed to NOx I could smell it almost everywhere for a few days.
Buses running on diesel smell specially strong. |
Yeah, I kept my windows open to ventilate and I remembered I could degassify by spraying bicarbonate solution. Which I did. my apartment is quite
clear now. But like I said, while these two synths weren't as bad as the previous times, I need to make absolutely that I have no more inhalation
going forward. I need to wait a while for the weather to get warmer before doing any more nitrations.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
OK so some final notes on this. While the final yield is obviously to be decided. I had to use a lot more water to crash out the picric acid because I
could not break up the chunk that was at the absolute bottom. One thing I needed to keep in mind for the future (if I ever do this again!) is that I
probably will need to add water to the acid BEFORE I put it in the fridge. Obviously the water must be ice cold and it must be added dropwise (for all
practical purposes) as putting the picric in the fridge caused it to solidify both times. But in the first time I was able to breka up the chunks
somewhat without excessive water. I was not able to do so in the second time in the end had to pour in boiling hot distilled water.
That's a note to self for the future.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
Wow. I dried out the batches separately. I am quite astounded. From my first 'hot' batch I measured out 315.60 grams from a starting 150 grams of
salicylic acid, 600ml sulfuric acid (h2so4) and 270 grams of KNO3 (potassium nitrate).
my 2nd batch of similar reagents but double the distilled water (around 1300ml instead of 800ml water) is 242.52 grams. This means I ended up with a
raw unrecrystalized total of 552.54 grams.
from 300 grams of salicylic acid this is a total of 1.8418 grams total. Even Rosco only made a total of 1.2g surplus, not 1.84. Then again, he started
from acetylsalicylic acid, and not salicylic acid.
I am both proud and terrified of myself! Going to report the total weight after recrystalization.
[Edited on 25-2-2023 by ManyInterests]
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
I got one question... I got a lot of left over liquid (which I will boil mostly down, but leave only a little left since I am confident that the
remainder will have whatever is left of the sulfuric acid in it) but after boiling it down, and even when cooling down slowly, the picric has a
reddish tint to it.
Is this a problem? did it degrade or something happen to it? I need to mention that in my previous synths using ASA There was a lot of blobs of
impurities at the bottom, but in this circumstance, I find nothing. I believe this cold mean that I have some extremely pure picric acid right off the
bat. But a recrystalization is always a good idea, if not to get some better crystal shapes.
But I am worried about the reddish tint to everything. Why does that happen? I know that when you dissolve picric acid in hot water the color of
everything turns red, but is that an issue that I should be concerned about?
 
|
|
|
B(a)P
International Hazard
Posts: 1139
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
| Quote: Originally posted by ManyInterests | I got one question... I got a lot of left over liquid (which I will boil mostly down, but leave only a little left since I am confident that the
remainder will have whatever is left of the sulfuric acid in it) but after boiling it down, and even when cooling down slowly, the picric has a
reddish tint to it.
Is this a problem? did it degrade or something happen to it? I need to mention that in my previous synths using ASA There was a lot of blobs of
impurities at the bottom, but in this circumstance, I find nothing. I believe this cold mean that I have some extremely pure picric acid right off the
bat. But a recrystalization is always a good idea, if not to get some better crystal shapes.
But I am worried about the reddish tint to everything. Why does that happen? I know that when you dissolve picric acid in hot water the color of
everything turns red, but is that an issue that I should be concerned about? |
I assume you are not trying for mass production here? The whole idea of taking the liquid off your crude crystals is to get rid of all of your
impurities. Your liquid will contain all sorts of impurities and is best neutralised and discarded. If you chilled your initial reaction mix right
down you are likely to be disposing of no more than several g of picric acid per litre. Best to focus on your crop of crude crystals and getting them
purified. Trying to increase yield by extracting product from your waste solution is of no real value unless the desired product is high value or you
are trying to optimise an industrial process. With energetics in particular obtaining a pure product is very important. You don't want to be
introducing unknowns.
Edit - typo
[Edited on 26-2-2023 by B(a)P]
|
|
|
Herr Haber
International Hazard
Posts: 1236
Registered: 29-1-2016
Member Is Offline
Mood: No Mood
|
|
It worries me that you synthesized such an amount. You dont need that much for proof of concept.
I agree with B(a)P, focus on purifying what you have. As for getting most out of the mother liquor, I'd say you reach a point quite fast where you
have to invest a huge amount of time / energy for diminishing results. I'm not saying dont to it, it's educative and you can get interesting shapes
from those crystals. But after doing it once and weighting the results you probably want to focus your efforts on your goal.
I reached this same conclusion with silver refining. I spend an awful amount of time to get all the silver out of solution. Litterally miligrams...
And then when I spill the same amount on the table I just wipe it off with a sponge...
If you're worried about your PA still having some impurities just recrystallize it once more. Do the maths: how much PA you have, how much water
you'll need, how much PA will be lost still dissolved in the water at room temperature ?
Not enough for you to care about it.
But man... I'd never want to recrystallize that much with just water. Is IPA / ethanol / methanol available for cheap where you are ? All would work
as co-solvents. As for the ratios... well... eyeballing works as long as you've kept enough volume in your container to add more alcohol.
Hot filter, let cool to room temperature then filter out the PA. No need for a final wash. Drying should be faster than with plain water. I see in
your picture that your PA is a bit damp. You can squeeze out water when picking it up with your disposable gloves. BE CAREFUL ! Dont break the filter
Important note: if you use a co-solvent as I suggested PA form will be different than from water. IPA is cheap, some people dont recommend it because
it makes platelets / scales that are "hard" to pour and to pack for maximum density.
Sieving through a fine plastic strainer will get you powder. Wear a mask while doing this or enjoy the bitterness of PA in your throat
The spirit of adventure was upon me. Having nitric acid and copper, I had only to learn what the words 'act upon' meant. - Ira Remsen
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
| Quote: | It worries me that you synthesized such an amount. You dont need that much for proof of concept.
I agree with B(a)P, focus on purifying what you have. As for getting most out of the mother liquor, I'd say you reach a point quite fast where you
have to invest a huge amount of time / energy for diminishing results. I'm not saying dont to it, it's educative and you can get interesting shapes
from those crystals. But after doing it once and weighting the results you probably want to focus your efforts on your goal. |
I know how you feel. I had no idea I would end up with this much. My guess is that the Salicylic acid is so much better than the ASA that I used that
I ended up with much, much more than I ever thought I would end up with. Most of the picric will end up in a bonfire when I am done with all this
because I really don't need that much. I know it burns well but will not explode, which is important. Like I said, when I used ASA I didn't end up
with this much surplus. I usually got roughly around what I put in, or even less (and that's only the crude yield. After recrytalization I ended up
with far less as well). If I had known I would have gotten 1.84 grams per 1 gram of salicylic acid I would have done just one synth of 100 grams of
salicylic as a 'final exam' and called it a day. I seriously underestimated what my final result would be, and it was likely due to the ASA I was
using probably wasn't as pure as I thought it was. The salicylic acid I got? That stuff is as pure as it gets.
The mother liquor is gone. The stuff I will be boiling down is the liquid I used to recrystalize, but I will be leaving behind a few hundred ml to
make sure that any left over H2SO4 is gone and ditto for any other impurities.
IPA and methanol are super cheap, and I have a good supply of 95% ethanol, too. For using water this stuff takes HOURS. I use 2100ml of boiling hot
water (I preboil it before pouring it in the beaker) and then start the heating. Since it is already hot the heating won't be necessary. It takes
around 4.5 or 5 hours to make it down to 700ml or so before I take it off the heat.
The picric is damp and I am willing to give it a squeeze. I learned that when filtering it I need to use 4 coffee filters on top of one another in
order to make sure the filter is strong enough for a good squeeze with breaking. And I always use glove. I have lots and lots of disposable nitrile
and vinyl gloves available. ditto for masks. I have lots of surgical masks to use in addition to my gas mask.
And I said it before and I will say it again. I am done making picric acid. I think after making all of this I have proved to myself that I can make
it. I need to move onto something else!  To be honest after this, most
energetics making is starting to get repetitive and I might even retire from chemistry for a good long time. I still got other journeys to go on in
energetics making, but those are for another time and another topic! To be honest after this, most
energetics making is starting to get repetitive and I might even retire from chemistry for a good long time. I still got other journeys to go on in
energetics making, but those are for another time and another topic!
Edit: To a large extent seeing my yield made me think of the video from WessonSmith (the NHN guy who doesn't post here anymore) who has a video where
he made what appeared to be dozens of grams (if not over 100) of NHN. That was... heart stopping!)
[Edited on 28-2-2023 by ManyInterests]
[Edited on 28-2-2023 by ManyInterests]
[Edited on 28-2-2023 by ManyInterests]
|
|
|
Texium
|
Thread Topped
28-2-2023 at 10:27 |
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
Thanks Texium! I basically made this post a sticky!
That being said, I have recrystalized everything and I am very glad I did. While I tried to 'reclaim' more picric from the water I used to
recrystalize it by boiling that stuff down... but I decided against it. I saw some shimmering in the water that, to me, was indicative of sulfuric
acid that was still left over, and I decided that it was best to get rid of it. While I didn't squeeze out all the water from the recrystalized
picric, the paper towels (of which I will need to replace frequently) should get rid of any remaining unwanted sulfuric acid. I neturalized everything
with sodium bicarbonate (also got rid of the little bit of picric that remained) and poured it down the drain.
Also the redness appears to be gone. Maybe it was a temporary thing, but either way the color is more yellow now. I hope that now I have pure picric
acid to turn into ammonium picrate. I really hope they are some good TNP this time.
[Edited on 1-3-2023 by ManyInterests]
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
I am back at this because now I finally got around to making my own ammonia solution. After some mishaps (like creating a saturated solution and
flooding my apartment with ammonia fumes!) I managed to make 1000ml of 24% ammonia solution. I feel very proud of my achievement. Next time I hope to
hit the 28% concentration.
But that being said, when it comes to making ammonium picrate, as I have said previously, I know an excess of ammonia solution is needed. I have no
problem dropping in a few hundred ml of this, but how much is exactly needed? like if I wanted to turn 100 grams of picric acid into ammonium picrate,
would I need 150 grams of the ammonia? around 600ml of my supply?
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
Since I cannot edit my last post I'll make a new one.
So I learned from Urbanski that there are two ways of making ammonium picrate. The first is with ammonia water (or as mentioned by XeonTheMGPony,
bubbling ammonia gas directly into the solution), and taking a measure of the pH. Since picric acid is... well, an acid, once the acidity level starts
to drop to a certain point, it should be all ammonium picrate at that point. It doesn't even need to be a very strong base, but around 9 or 10 is
sufficient.
A second method and I learned this from Urbanski is... to use ammonium carbonate. I never thought of that, but if using sodium carbonate/bicarbonate
will yield sodium picrate then... yes, it does stand to reason that neutralizing it with ammonium carbonate will yield ammonium picrate. Again, it is
all about measuring the pH here.
I am thinking of getting some ammonium carbonate just to try that method out.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
OK, I used 77.5 grams of picric acid in around 1400ml of boiling hot distilled water. I started adding ammonium carbonate. I did not count how much
carbonate I put in, but I stopped to take a pH reading and I was slightly over 9, but under 10. I didn't realize that the solution would still be
fizzing even it reached alkaline levels.
I let it fully cool in the fridge after this and I filtered out the very nice golden yellow needle like crystals that are characteristic of yellow
ammonium picrate. I still had 1200ml of liquid. I calculate based on the soubility of ammonium picrate that there should be around 8 grams of dunnite
in the liquid. Right now I've evaporated the liquid at 90 Celsius until it is around 500ml. I think once it fully cools down, it should be around
400ml or so. I hope to salvage at least half those grams.
|
|
|
ManyInterests
National Hazard
Posts: 930
Registered: 19-5-2019
Member Is Offline
|
|
OK the first attempt produced good quality ammonium picrate, but I should have dried to dry all the liquid to full dryness to pull out as many
crystals as possible, as I only got around 41.5g of dunnite from the whole thing,
I did a 2nd attempt with another 77.5g (this time using the 'old' triple recrystalized picric acid that I had handy. I decided to do it with a 10%
solution of ammonia instead of ammonium carbonate. I added 99ml of 10% solution, at first I put in only around 10 or 15ml, but I saw it immediately
turning red, so I decided to jump dump the whole graduated cylinder all at once.
The pH was a good 9 right off the bat, and it was VERY red. Urbanski was right in that regard. Ammonia water is much more likely to make the red
version of dunnite than yellow. It is a lesser quality, but just as powerful. I heard conflicting reports that the yellow is harder to set off, but
others that say that the red is harder to set off. Either way I am gonna mix them together in the end of the day.
I will not air dry my picrate, I will dry it over a hot water bath, this is not only faster, but I also want to get as much yield as possible from
this. I worked hard to get that picric acid and I don't want any of it going to waste!
Edit: It might not be the red version, I am seeing some yellow bits coming out of solution, but that could be yellow ammonium picrate and not picric
acid. It could be picric, too, but I am not sure, the pH was quite strongly base.
Edit2: Nope, it isn't red ammonium picrate, it is the yellow version! Good!
[Edited on 4-7-2023 by ManyInterests]
[Edited on 4-7-2023 by ManyInterests]
[Edited on 4-7-2023 by ManyInterests]

|
|
|
axd1995
Harmless
Posts: 6
Registered: 9-8-2023
Member Is Offline
Mood: No Mood
|
|
I used 20ml of 92% sulfuric acid and 3.6g of salicylic acid. After heating, I added salicylic acid, but it couldn't completely dissolve. I raised the
heating temperature and there was a strong phenol odor, but it still couldn't completely dissolve. Why?
|
|
|
B(a)P
International Hazard
Posts: 1139
Registered: 29-9-2019
Member Is Offline
Mood: Festive
|
|
| Quote: Originally posted by axd1995 | | I used 20ml of 92% sulfuric acid and 3.6g of salicylic acid. After heating, I added salicylic acid, but it couldn't completely dissolve. I raised the
heating temperature and there was a strong phenol odor, but it still couldn't completely dissolve. Why? |
How hot did you go? You need to get to 115 C to get it to fully sulfonated. It sounds like you have enough sulfuric acid, so my guess is you did not
go hot enough.
|
|
|
DennyDevHE77
Hazard to Others
Posts: 167
Registered: 15-9-2014
Member Is Offline
Mood: No Mood
|
|
Hello everyone, there is a recipe for the preparation of picric acid from sulphanilic acid without the use of concentrated sulphuric acid.
If anyone is not aware, I will give the methodology written out of the book "Laboratory preparation of explosives" by Solonin A.A..
"30g of sulfanilic acid is shaken with 250 cm³ of water and with 12g of sodium salt of nitrous acid. After careful stirring, carefully under strong
cooling with ice water, about 100 cm³ of the cooled solution of 10 grams of sulphuric acid of specific gravity 1.84 g/cm³ in 100 cm³ of water is
poured in.
After adding the sulphuric acid while cooling, the mixture is stirred from time to time for one or two hours, and left to stand for two hours at
ordinary temperature. The obtained porous mass is filtered on a Buechner funnel, transferred into a beaker and then, with stirring and heating on a
water bath poured 96g of nitric acid of specific gravity 1.38 g/cm³.
At the end of pouring nitric acid, heating is carried out until the end of gas emission and then left to stand for a day.
The released picric acid is filtered off on a Buechner funnel, washed with cold water."
There is a scheme of reactions (on the picture)
I was attracted to this method because sulfanilic acid costs about the same as phenol, and in general all substances are legal and easily available.
Also, I'm currently experiencing some problems with concentrated sulfuric acid. But I still haven't found correct information on the yield. And also
the exact temperatures are not specified, and I think there may be pitfalls here.
If someone has experience in synthesis of picric acid by this method, please share your experience, thanks in advance. Personally, in general, this
method seems to me to work, but I do not expect large yields, as the molecule of sulfanilic acid is already heavy enough.
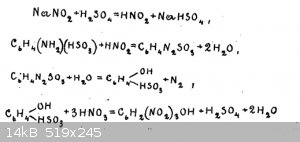
[Edited on 17-8-2023 by DennyDevHE77]
|
|
|
| Pages:
1
..
28
29
30
31 |








