| Pages:
1
2
3
4
5 |
LSD25
Hazard to Others

Posts: 239
Registered: 29-11-2007
Member Is Offline
Mood: Psychotic (Who said that? I know you're there...)
|
|
Yeah, this shit is cursed - I have been breaking glassware like it's free (problem being it fucking ain't) every FUCKING time. I 'think' the crude
product from dry NH3 gas is not too bad purity wise - it doesn't seem anywhere near as prone to autooxidation and unusual products for a start. What
suprised the fuck out of me is the sheer difficulty encountered gassing the amine - I passed fucking shitloads of dry HCl(g) into this crap, while
cold, there was nothing - no colour change, no precipitation, nothing, just clouds of HCl (which is right up there in my list of things I don't like).
After doing this, I then tried heating the shit to see if I could get a crystaline product - well, something, but each time it was heated more and
more HCl gas was given off (obviously held by the Acetone, as one would expect - but if so, why the fuck didn't it react with the amine?).
I was thinking of trying H3PO4 (I have serious problems getting access to conc. H2SO4 (I think it was thrown on peoples faces here once) - I'll make
it nice and fresh from polyphosphoric acid) - there is a couple of interesting things to report - the contaminants when I washed the broken glass with
EtOH (95%) precipitated when I washed the glass with water in the funnel (obviously not too soluble in less than 90% EtOH), leaving a very pale yellow
solution - which I have yet to process.
Seriously, I am really getting the shits with this stuff. How much do you need? Isn't it usually used in catalytic quantities only?
PS When heating the Actone/TAA/TAA.HCl solution, there was the separation of a decent amount of yellow oil, as this was heated further some of it
evaporated and some changed to a reddish oil which finally went black and tarry (I'm guessing this is the n-oxide? which suggests autooxidation).
One thing I was thinking of, is the 2,2,6,6-tetramethylpyridine more stable than TAA? If so, save trying to isolate the sucker, how about adding some
phosphonic acid and some iodine and just isolate the basic amine at the end? Stripping off the excess acetone first would greatly assist in this
endeavour, but I think HI is probably going to be more than enough to desoxygenate the 4-keto position?
[Edited on 16-5-2008 by LSD25]
Whhhoooppps, that sure didn't work
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Considering the impurities present with the TAA direct oxydation of the crude TAA gives a very dirty product. even the oxydation on the distilled
freebase gave a very complex mixture of products. But there no evidence this would prevent the 4-oxo-TEMPO present from doing it's job, but you will
introduce alot of impurities.
I find it better, when trying out a somewhat new procedure, to get, at least at first, a pur product, and to be sure the problems arising are not du
to impurities present. Using a crude product poses to much variables to be sure it works or not.
But once pur 4-oxo-TEMPO has been isolated, and proved to be efficient in catalysing oxidations, the crude product could be tried, if similar
results are obtained, it might not be worth the effort of isolating pur intermediates/catalyst. But for now, let's do it cleanly 
Synthesis of 4-oxo-TEMPO according to US 3,959,295

first step:
In a 250mL 4-neck RBF, equipped with a gas inlet, a condenser, a thermometer and magnetic stirring, 25mL (340 mmol) of acetone and 9mL of MeOh were
charged, followed by 1.1g (20.56 mmol) of freshly fused NH4Cl.


A 500mL erlenmeyer was charged with ~100g of humid, crude NH4Cl (byproduct from hexamine hydrolysis), containing roughly 80g (1.5 mol) of NH4Cl,
and a stir bar. The erlenmeyer was fitted with a 250mL addition funnel, containing 150mL of 40% w/v NaOH solution (1.5 mol). A gas outlet was
connected to the top of the addition funnel, linked to a wash bottle containing solid NaOH pebbles, itself connected to the gas inlet of the RBF.

The NaOH solution was slowly dripped onto the NH4Cl, generating a constant, easily monitered flow of NH3. the acetone/MeOH was gassed at
1bubbles/sec, with vigorous stirring, at room temp (17°C). A latex glove was attached to the top of the condenser, keeping in any unabosrbed NH3. It
was not "blown up".
Gassing was continued for 3H, until the mixture was saturated. Near the end, the temp rosed over 30°C, so an ice bath was applied to keep temp
under 15°C. Once saturation was definatively attained, the gas inlet was removed.
Second step
75ml (1.02 mol) of acetone are then added, turning the whole mixture milky white as a fine solid precipitates, and agglomerates upon stirring.


The flask is then heated to 50°c with an oil bath. After a few minutes, the solid dissolves completly, offering a water-clear solution. After1 hours
stirring, the mixtures takes on a yellow colour, and afetr 15H a dark red solution is obtained.


The condenser is replaced by a fractionnal distn setup, with a 150mm vigreux, and the gas tube connect to the vacuum inlet and immersed in a dilute
H2So4 solution. Heating is increased, and the excess acetone removed. After ~75mL are distd, slight vacuum is applied.

A dark red, viscous residu is obtained. It fumes when vacuum is disconnceted.

2.25mL (125 mmol) of water are added to the cooled residu, and the flask is immersed in a ice water bath with slow stirring. After 1 hour, there still
isn't any solid forming. apparently the product is too impure to be able too form a hydrate directly.
The residu is then titrated with conc. HCl. Over 20mL are required until acidic to dampened pH-paper. 100mL of IPA are added, and distn under vacuum
at 60°C. this is renewed with 50mL IPA. Unfortunaly, the distn was left unattended for too long, and the residu heated up considerable when the IPA
was completly removed, but not for long.
The thick black/red syrup was cooled down, thickening alot, and 50mL toluene added. A semi-solid tarry goop is formed, which doesn't solidify when a
little sample is triturated under acetone, although it looks likes a cristalline paste, it stays liquid.
30mL acetone are added to the two layers, the solvent top layer taking ona red color, without much effect on the tar. 30mL of IPA are added, which
dissolves most of the tar, forming a black/brown homogeneous layer, and a white/yellow solid sperates out immediatly. This is left to sit for 30min,
then vacuum filtered. A yellowish powder is obtained, which is generously washed with acetone, and pet ether before being dried by suction, giving a
very light, fluffy beige poxder.


A total of 10.7g (55.87 mmol) of triacetoneamine hydrochloride is obtained. The solid will be recrystallized with the old sample, from IPA/acetone,
or IPA/pet ether.

Comments
The workup is the problem here. The cristallization is far from optimal. The filtrate has been placed in the freezer to see if more product will
cristallize, but next time I will rather try adding conc. H2SO4 to the reaction medium directly as advised by the patent, or adding ethanolic oxalic
acid to the residu, as advised in some of the articles KMnO4 very kindly posted.
The oxalate salt seems pretty easily isolated, and fairly stable.
It's a pity the hydrate didn't readibly form, this would definatively be th ebest way of stocking TAA for short to medium periods. No need of
basifying before oxydation. I guess it could be kept in the fridge for longer stocking time, but as it is commercially avaible under this form, I
guess it's pretty stable.
In any case, this seems to be a good reaction: no need of gassing for several consecutive days, waiting a week, etc. Apparently the methanol does a
good job at dissolving alto of NH3, as pur acetone doesn't dissolve much by itself, hence the consecutive gassings in sevral articles. The gassing is
pretty staright forward and doesn't require constant presence once stable. Just gas the shit out of it until saturated  . I think a little less acetone could be added in the second step, I followed the
second series of examples in the patent, but the first one used 1/3 the amount of acetone in the second step. . I think a little less acetone could be added in the second step, I followed the
second series of examples in the patent, but the first one used 1/3 the amount of acetone in the second step.
Considering the ease of the reaction it would be adviseable to do it at a bigger scale. If the TAA can be directly isolated from the reaction medium
by conc. H2SO4 as claimed by the patent, no need of distilling the excess acetone, which saves quite some time. The ~8.6g of freebase should yield
more than enough 4-oxo-TEMPO, but I might just try another reaction with using either H2SO4 or oxalic acid to isolate the amine.
Once thta's done, oxidation time!
Come people, jump in! This is easy and rather a break through in home-chemistry: a 100$/gram catalyst ( !! ) easily synthesized from scratch!
The second part, checking out the efficienty and range of use of this catalyst is going to be a pretty large project too.... Obviously, i will be
starting with benzylic alcohols to aldehdyes oxidations, becasue that's why I decided on try to make the catalyst in the first place, but it can also
be used for aliphatic aldehdyes, nitriles from amines, etc
Here is the patent for the lazy people  : :
[Edited on 23-5-2008 by Klute]
Attachment: US 3959295.pdf (264kB)
This file has been downloaded 1336 times
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
detritus
Harmless
Posts: 19
Registered: 27-4-2008
Member Is Offline
Mood: No Mood
|
|
i did see one patent that did reactions on the bisulfite addition product of TAA. i will try to dig it up. but it gives some confidence that it
would actually form an addition product
|
|
|
kmno4
International Hazard
Posts: 1497
Registered: 1-6-2005
Location: Silly, stupid country
Member Is Offline
Mood: No Mood
|
|
Nice... I have always wanted to do this, but I am too lazy
Are you able to check up melting point of your product, to confirm (let's say) it is indeed triacetonamine hydrochloride ? Aldrich gives 198 °C.
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I will do so after recrysatllization, I think there must be a little diacetoneamine and other uncyclized amines in there too.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
chemoleo
Biochemicus Energeticus
Posts: 3005
Registered: 23-7-2003
Location: England Germany
Member Is Offline
Mood: crystalline
|
|
Synthesis of the acetonine nitroxide radical
Will be of interest to most here...
Title: Synthesis of acetonine nitroxide radical and 1-hydroxy-2,2,4,6,6-pentamethyl-1,2,5,6-tetrahydropyrimidine
Tetrahedron Letters 41 (2000) 179–181
Attachment: acetonine nitroxide radical TEMPO.pdf (76kB)
This file has been downloaded 2263 times
Never Stop to Begin, and Never Begin to Stop...
Tolerance is good. But not with the intolerant! (Wilhelm Busch)
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you very much Chemoleo! Indeed, it is pretty interesting!
That finally gives us an idea of the product isolated when the recation is done at room temp (its a pity they don't give the physical properties of
the acetonine, surely a yellow oil?),b ut the fact it also forms a satble nitrosyl radical means that the crude distillate I oxidized a month ago can
act as a catalyst even though it doesn't contain only 4-oxo-TEMPO.
As they claim the acetonine form TAA upon reaction with acetone and ammonium/calcium chloride, I guess this si what happens in the second step of
the reaction.
But it's pretty hard to say what exactly is produced when reaction acetone and ammonia at room temp: some say high yileds of diacetoneamine are
produced, they say the aectonine is produced in high yields... Surely a complex mixture of boths (and others). The fact that DAA and this acetonine
both form salts would suggest that the less recent preparations of DAA would give a product contaminated with the acetonine.
So i guess preparation involving a rather long reflux with excess acetone would be the more appropriate to obtain TAA with the less contamination
possible, and adding a stronger lewis acid in the second stage might be beneficial (zinc chloride seems like a good suggestion from the article).
If using the catalyst straight as is, minor contamination with acetonine radical shouldn't be a problem, but it will when trying to derivate the
4-oxo-TEMPO to a solid catalyst.
For the next preparation of TAA, i will try out more elaborate conditions: distilling the acetone over CaCl2 or similar, using recrystallized, fused
NH4CL as catalyst, and a more pur grade of NH4Cl to generate the ammonia.
I'm going to request the preparation of TAA referenced in this article, that 90% seems interesting and might containa detail that could greatly
help.
EDIT: possible bad news: in patent US6608220, bisnoralcohol is converted to bisnoraldehdye by using NaOCl with catalytic Br- buffered by bicarbonate,
and a derivative of TEMPO. They use 4-hydroxy-TEMPO in most case, but give no yields, conversion or selectivity, just "the compound is obtained".
On the other hand, example 6 is more worrying:
| Quote: |
Bisnoralcohol (I) to Bisnoraldehyde (II) with 4-oxo-TEMPO
A mixture of bisnoralcohol (I, 6.6 g), 4-oxo-2,2,6,6-tetramethylpiperidine-1-oxyl (18 mg), dichloromethane (30 ml), sodium bicarbonate (180 mg),
potassium bromide (238 mg) and water (5 ml) is cooled to 1°. Then aqueous sodium hypochlorite (14.6%, 11.4 ml) is added to the mixture over a 15 min
period. The reaction produced the title compound but in only a 7% conversion of bisnoralcohol with 58% selectivity for bisnoraldehyde.
|
But this odesn't mean 4-oxo-TEMPO is much less efective in catalyzing oxydation of primary alcohols to aldehydes:
First, they add the hypochlorite in 15 in that example, where as they add over 5 h in the other,
Secondly, they oxidize a pretty large and delicate molecule.
The only difference I can think of between the oxo and hydroxy-TEMPO is that the oxonium is slight more stable with the oxo- than with the hydroxy-,
though seperated from the nitrogen by two more carbons... But obviosuly there could be lots of other things to consider.
I intend on preparing 4-hydroxy-TEMPO in any case, and compare it efficienty with the oxo-, and also try making methox-TEMPO and ethoxy-TEMPO, as
apparently the hydroxy-TEMPO is very easily alkylated.
EDIT 2 :
I really owe KMnO4 : he posted a series of articles on TEMPO derivatives in the ref forum, including a very complete review on hindered
piperidines, which really give more perspective of modification of 4-oxo-TEMPO, there are litteraly tons of way of modifying TAA in different ways..
He aslo joined a excellent article detailing the oxidation of TAA, with H2O2/WO4 2- and H2O2/CO3 2-, indentifying all the impurities and their
caracteristics (even the Rf's! ) and presenting a remarkable workup for the
H2O2/CO3 2- (which is much cleaner than the WO4 2-! I wonder why they even want to use versanate and tungsate salts when carbonate works better?! ),
that I am attaching because it's so usefull.
Here is the workup:
| Quote: |
TAA oxidation in the H20,/Na2C03 system (the modification of the procedure
TAA (20 g, 0.129 mol) is added with stirring and external cooling (cold water) to a solution of anhydrous sodium carbonate (41.7g, 0.393 mol) and
300,:, hydrogen peroxide (200 g, 180 cm5, 1.765 mol). The stirring is continued for two days. Sodium chloride (about 60 g) is added, and the mixture
is left overnight in a refrigerator. Red crystals (16.6 g) are filtered off and dried over silica gel in a vacuum dessicator. The crystals are
dissolved in 600cm3 of n-hexane and the residue of sodium chloride is filtered off. After evaporation of the solvent, 16.0g (73%) of TAAO (m.p.
35-37°C) is obtained. The impurities 2-4 are not detected (TLC).
The elementar analysis: calculated for C9H,,N02 C 63.50, H 9.47. K 8.23; found C 63.60, H 9.6'5, N 8.22.
The m.s. is the same as in [I.].
|
I figured out I could simply use the HCl salt in conc. aq solution, and use 1 more eq of Na2CO3, to directly freebase the TAA s it is added, the NaCl
produced won't be problematic as more is added after.. I am very happily surprised that the 4-oxo-TEMPO seperates out as crystals so easily! that
saves alot of purification steps! But I guess very pur TAA should be used, which involves at least 2 recristn.
Unfortunaly, the silica/SO2Cl2 article showed that this preparation doesn't give a sulfonly chloride functionlized silica, but a sulfate ligand, with
two Si-O-S bonds, so no free acid to attach a TEMPO to... Both S-Cl bonds react with a -OH group.
So chlorosulfonic acid is the way to go, but I don't think i will trying this until I get a few derivatives of 4-oxo-TEMPO.
One good news from the rview is that the nitrosyl isn't affected by NaBH4 reduction! That means 4-Oxo-TEMPO can directly be reduced to 4-OH-TEMPO. It
might be wiser to start off reducing TAA, and comparing the obtaiend products to see which is the purest. I haveyet to find a procedure detailing the
reaction conditions of such a reduction,, I'm not sure fi it would be better to to a usually or reversed addition. I was thinking of adding the TAA in
AcOEt to an excess of NaBH4 in MeOH or EtOH.. If anyone know of such a procedure (i checked for reductions of piperidones in general, an didn't find
anything specific).
4-amino-TEMPO is easily prepared from 4-oxo-TEMPO: reaction of 4-oxo-TEMPO with hydroxylamine, and reduction of the oxime, and the review also
explicites that Wolf-Kishner reduction of 4-oxo-TEMPO proceeds easily to TEMPO, if anyone wants to mess with hydrazine...
Lots of things in perspective
[Edited on 25-5-2008 by Klute]
[Edited on 25-5-2008 by Klute]
Attachment: fulltext1.pdf (233kB)
This file has been downloaded 1434 times
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I would like to add that MeOH is a excellent recrysatllization solvent for TAA.HCl, a dual solvent with MeOH/acetone yields nice small needles,
colorless to yellow. Pictures will come tomorow.
IPA seems to dissolves only very little product even at boiling temperatures:
10.7g of TAA.HCl were placed in a 250mL erlenmeyer, and 50mL IPA added in portions and heated to boiling on a hot plate. after 10min, hardly any
beige poxder had dissolved, although the IPA had takena slightly yellow colour from impurities (an IPA boil could be a good idea to purify crude
TAA.HCl ).
MeOH was then gradually added. 13mL were added until complete dissolution, giving a ratehr dark orange solution. Thsi was left to cool, and seeded
with a grain of crude TAA.HCl, which initaite cristn immediatly. Small needles started to form.
After 30min, 20mL acetone:pet ether 2:1 was very slowly added from the sides of the erlen, causing cristn on the surface at first. The erlen was
left to rest for another 30min, covered, then sealed and placed in the fridge for 3H, then in the freezer overnight.
The crystals will be filtered tomorow; and rinced with a little cold acetone:pet ether.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Bolt
Hazard to Others
Posts: 188
Registered: 26-3-2007
Member Is Offline
Mood: No Mood
|
|
Klute, thank you for your dedication; hard work; and illustrated, detailed writeups on this project!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
My pleasure Bolt! :) This project is really catching me on! I will not rest until TEMPO catalysts are found in every home-lab :D
Oxydation of triacetoneamine with H2O2/K2CO3
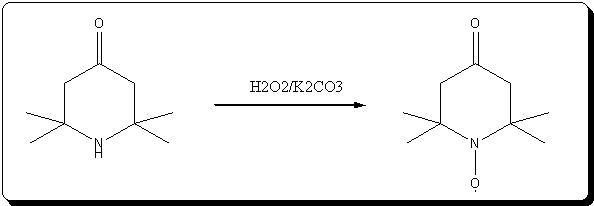
This is a small variation of the methode described in the article KMnO4 retreived lately, that i attached just above.
A made a few modifications, and could have done things a little better; but at least know I know what I will do next time :)
In the procedure, they use anhydrous Na2CO3. At home, I had the choice between very technical (hardware garde) hydrated Na2CO3 (unknown hydratation,
less than decahydrate, and of questionnable purity, possibly containing soaps/detergents), and anhydrous 98% K2CO3, so I prefered using K2CO3. It
should be noted that in most procedures using H2O2/CO3 2-, the sodium counter ion is used. In the patents, they claim potassium carbonate can used,
and a example gives a very high selectivity, so it shouldn't be a problem.
Procedure
In a 250mL wide neck erlenmeyer, 21.8g (157.73 mmol, ~3.4eq) were dissolved in 64.35mL (631.02 mmol; 13.7 eq), 30% H2O2. This caused some warming up
and fizzing. The milky solution was cooled in a ice bath with slow magnetic stirring.

8.83g (46.12 mmol) of recrystallized TAA.HCl were crushed into a finer powder, and added in portions to the solution. AT first, nothing much seemed
to happen, no CO2 evolution, the powder jsut formed a suspension.

1.88g (47,00 mmol) of Naoh were weighed and added as a solid in small portions to the suspension. The solids seemed to increase in volume, and
whiten. A thick slurry of a milky white fluffy solid was formed: the TAA formed a hydrate as soon as it was based. As O2 bubbles were evolved, the
voluminous solid formed a foam, hard to stir in surface. It was broken down regularily with a thermometer, to facilitate magnetic stirring.



A limpid colorless liquid was under the layer of foam, looking alot like egg's white that has been "beaten" to snow (don't know the expression in
english, you knwo what I mean :) ). It was continuously stirred with the thermometer to mix up the foam.
the temperature was left to climb up to 20-25°C, sometimes up to 30°C, but kept under that temperature by applyling a ice bath now and then. There
wasn't any strong exothermic reaction, the heating was due in part to the hotplate used to stir. There was continuous, but gentle, evolution of
bubbles.
After 4h, the liquid started to turn limpid yellow, and the amount of solid/foam seemed to diminish. Stirring was much more easier.



At 6H total reaction time, the white foam had turned to light orange cristallin solids floating on the surafce of the liquid; the temperature ahd
increased over 40°C at one point, and was quickly cooled donw to 15°C with the ice bath. There was still noticeable bubbling.

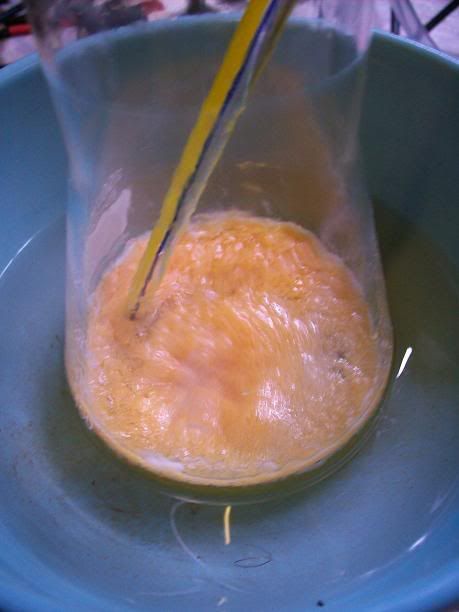
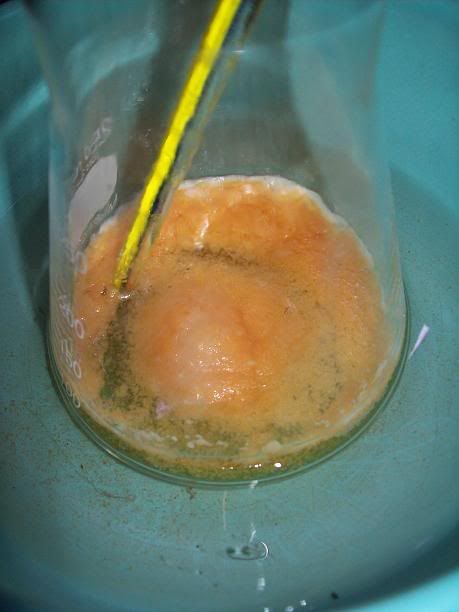
After 20H, a dark orange/red semi-solid tar like gum is obtained. No more bubbling can be seen.
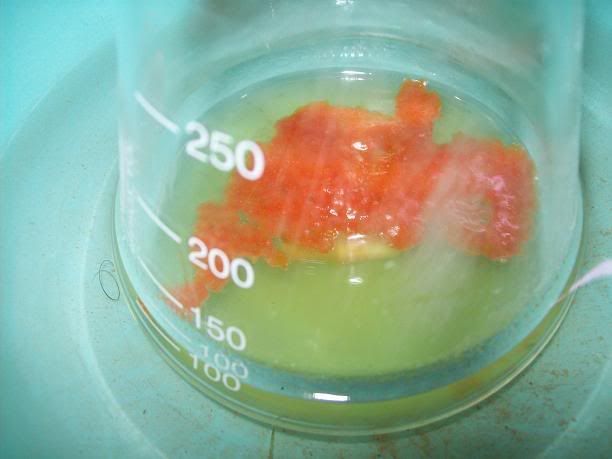
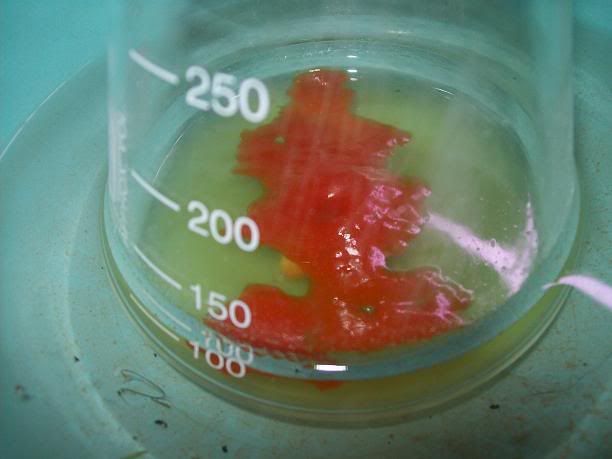
I'm thinking starting the work up tonight, as it doesn't seem to evolve that much now. I hope the semi-solid will solidify entirely whne refrigerated,
to be able to filter it effectively, otherwise I will have to do a solvent extraction. In that case, I will neutralize with acetic acid until I have a
pH of ~4, so that any unrecated TAA will stay in solution and the nitrosyl radical will not be degraded.
Comments
All the glassware was washed with demineralized water to avoid contamination with metal salts, which would promote the decomposition of the H2O2 (note
the huge excess).
The reagents were added in a bad order: the H2O2 was added to the weighed K2CO3, which caused some lumps at the begginign, and possibly a little
decomposition from the basic conditions when only the first portion of H2O2 was added.
Apparently, potassium carbonate cannot freebase TAA.HCl. I will have the double check the pKa of TAA, but I don't recall it was that high... But, I
wasn'texpecting the formation of the hydrate, so the transition could be hard to notice with the carbonate.. If the amine was already freeabsed,
adding the NaOh would cause extra decomposition of the H2O2, as it is favored by high pH.
I should have: added stoechiometric amount of NaOH to TAA.HCl in minium water, then added the K2CO3/H2O2 solution to the freebase/hydrate (prepared
by adding the K2CO3 gradaully to the stirred and cooled H2O2). I think it is important to cool the recation at the beggining of the addition, as the
reaction could spark off and over heat if not controlled.
I'm not sure if the hydrate formation can be avoided if using not-so-cold temperatures, or adding the freebase to a concentrated carbonate solution.
Even if it isn't practical, it doesn't pose a big problem to stir, it's just that the layer of foam isn't well mixed with the aqueous if not stirred
by a rod to send it into the vortex.
Next time, i might try the bicarbonate procedure again, but use their workup which seems to be a breeze compared to extraction, washing evaporation
and recristn.
PS:
from US 5,431,901:
| Quote: |
A. 2,2,6,6-Tetraperdeuteromethyl-4-oxo-3,5tetradeuteropiperidine (Triacetoneamine-d16) (V)
A mixture of ammonium-d4 chloride (3.45 g, 0.06 mole), acetone-d6 (99.5 atom %D, 12.5 ml, 0.15 mole), anhydrous sodium carbonate (3.18 g, 0.03 mole)
and magnesium oxide (3.0 g) was added to a 250 ml round-bottomed flask. The flask was capped with a rubber septum and wired, then the reaction mixture
heated in an oil-bath at 50° C. for 3 days. After cooling, 20 ml of acetone was added to the reaction mixture and the resulting mixture was filtered.
The recovered solid was crushed into powder, washed with 15 ml of acetone and then filtered with suction filtration. The combined filtrates were
concentrated to dryness. The resulting red liquid (7.2516 g) was distilled under reduced pressure to obtain 4.7480 g (56.7%) of a bright yellow liquid
(b.p. (boiling point) 54°-55° C./1.9 mm Hg) that solidified when chilled in a dry ice/acetone bath. The solid product subsequently was used without
further purification. Recrystallization of an analytical sample from anhydrous diethyl ether yielded white crystals, mp. 57°-58° C. [lit. 58° C.];
IR(KBr, cm-1):3580(m), 3260(m), 2220(m), 1700(s), 1530(w), 1265(s), 1140(m), 1050(m), 930(w); 13 C-NMR (CDCl3): 31.03(m), 53.50(m), 54.88(s), 211.19
(s).
|
I guess this is basicly the procedure referenced in the zeolite absorption article, but using d-ammonium chloride instead of 15-ammonium sulfate....
No gaseous ammonia, or extended periods (well 3 days, but better than 3 month :) ). i will definatively try this out.
[Edited on 28-5-2008 by Klute]
[Edited on 28-5-2008 by Klute]
[Edited on 28-5-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Couldn't wait until tomorow: the first home made 4-oxo-TEMPO


It is still contaminated by TAA though... Will see tomorow how to proceed... I'm just to tired right now, between work at work, and work at home, I'm
doing well over 18H a day
I will post details after a good night's sleep!
The oxidation needs optimizing, or at least waiting 48h total.
I will follow the more recent patents next time.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I restarted a TAA synthesis, following the same procedure with dry ammonia, but at a double scale. Hopefully the yield will be increased by isolating
the TAA as a hydrosulfate or oxalate.
The oxidation didn't come out as expected. Apart from the product above, there was alot of unreacted TAA in the mixture. After adding more H2O2 and
leaving it to proceed for another 24H, only white solids appeared upon refrigeration. These could be either TAA hydrate, or the hydroxylamine. They
will be isolated and analysed by TLC..
If anyone really wants the details on the workup of th eoxidation, I can provide them, but I prefer trying out a new procedure as this one just seems
to fall a little under what was expected...
I will post details of the TAA synth when done. Hopefully I will be able to start a NaBH4 reduction of TAA tomorow.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Synthesis of 2,2,6,6-tetramethylpiperidin-4-one according to US 3,959,295, second try
This time I (nearly) followed the patent's procedure, and it's proposed workup with conc. H2SO4. Works like a charm!
First Step
In a 500mL 3-neck RBF, equiped with a condenser fitted with a latex glove fixed with an elastic, a gas inlet, a thermometer and magnetic stirring,
63.2mL (1.377 mol) of technical acetone and 15mL of MeOH were introduced, followed by 2.2g (41.12 mmol) of freshly recrystallized and dried NH4Cl and
1g CaCl2 pellets.
Roughly 150g (2.8 mol) of freshly recrystallized NH4Cl were charged in a 500mL flat bottom flask, with a stir bar. The flask was mounted with a 250mL
addition funnel, itself mounted to a gas outlet, connected to the gas inlet of the RBF in series with a solid NaOH washbottle.
A solution of 120g (3 mol) NaOH in 250mL dH2O was charged in the addtion funnel



The NaOH dripped onto the the NH4Cl, producing a slow, constant flow of NH3 gas. The RBF was immersed in a cold water bath, to which ice was
periodically added in order to keep the internal temp under 20°C, most of the time between 10 and 15°C.
the flat bottom flask was heated on a hot plate to acheive better desorption of the NH3.
Once all the NaOH solution had been added, the NH4Cl and CaCl2 had completly dissolved into the colorless solution.
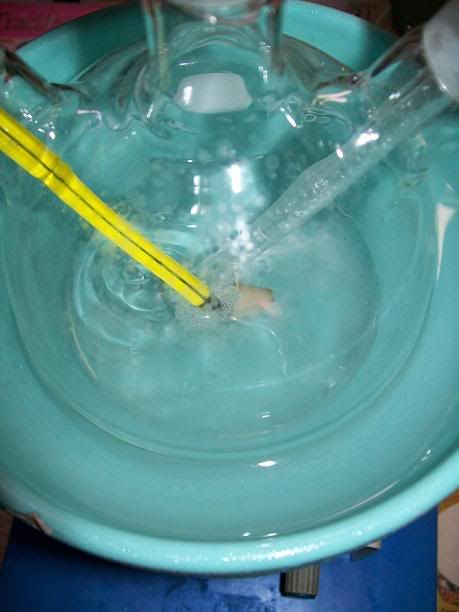
Increasing the heating of the flat bottom flask generated increasingly more NH3. Once the solution was nearly saturated (the glove started to get
blown up), 50mL of technical acetone were added to the flask's contents. The initially colorless solution immediatly turned milky white, and gradaully
cleared up as a white solid agglomerated onto the sides of the flask.


Gassing was continued as long as NH3 was generated. A very rapid flow of gas was now produced by the nearly boiling solution/slurry.
(Remark: In the first try, I dismantled the gas generator shortly after all the NaOH had been added. Apparently, a very large amount of ammonia
stays into the NaCl slurry, and extended heating is required for complete desorption. This must surely account for the low yield of the first
try.)
Once the flow of ammonia produced came to a near halt, the gas inlet was disconnected and immersed in a dilute H2SO4 solution. There was no suck back
at all.
The gassing took a total of 7H to proceed entirely. This period could be shortened by increasing the flow at the beggining of the gassing.
The solution was bubbling from ammonia (saturated) as it heated back up to 25°C (ambient), so a few tiny holes were put through the latex glove to
avoid excessive overpressure.

92.2mL of technical acetone were added (total amount of acetone introduced: 63.2 + 50 + 92.2 = 205.4mL; 4.477mol), without apparation of any
white solid, and another 1.1g (20.56 mmol) of NH4Cl and 1g of CaCl2 were added. The ammonium chloride dissolved immediatly, the CaCl2 did so more
slowly.

The colorless solution was stirred for 2h at room temp to avoid wastefull NH3 release. After that period, a small aqueous layer was noticed at the
bottom of the flask.

Second Step
The flask was then heated to 55°C in a oil bath. AS the flask heated up, there was only minimal NH3 release.
After 10H of heating, the solution had taken a light yellow colour.

Following the patent's procedure, 14g of H2So4 were charged into a addition funnel, and slow addition started. There was a extremly vigorous reaction
as the first drop touched the surface, generating alot of white fog and a strong sizzling noise.


The acid on the sides of the flask quickly turned to a white solid as it reacted with ammonia. White solids started appearing in the solution. The
addition was stopped, and it was decided to continue reflux without addition of acid.
heating was continued for another 5h, at which point the solution had taken a blood red colour.

The flask was cooled with a cold water bath, and maintained in a ice bath while conc. H2SO4 was slwoly dripped in. Again, the reaction was vigorous,
producing lots of white fog.

As the addition proceeded, the reaction became less intense, aswell as the generation of the fog. Any acid on the sides of the flask didn't not turn
to a solid as previously even after prolonged exposure.
The temperature was kept under 35°C by changing the ice bath regularly. 75mL IPA was added to thin up the slurry and help stirring. A thick red
slurry was formed. Once pH was acidic (to dampened universal pH paper, ~5), acid additon was stopped (over 25mL were required), and stirring at 10°C
was continued for 45min. The pH was checked at the end o fthe stirring period, and was found to still be acidic.
The thick slurry was vacuum filtered on a large buchner, taking some time to pass (the filtrate started boiling sevral times). The light bordeau cake
was washed with 200mL acetone, 75mL 2:1 IPA:Acetone, then 100mL pet ether (filtering was much easier after the first wash). This gave a off white
solid cake, and a blood red filtrate.


The filtrate placed in the freezer after been sealed with celophane, and the solid was dried by suction for 20min, then under a lamp.
73g of a light, free flowing off-white/beige powder was obtained.


The powder will be extracted/recrysatllized from methanol to remove ammonium sulfate and other impurities. It has a light smell of the free amine.
Comments
What a releif! the workup is definatively much simplier than assumed, and avoids lengthy and destructive distillation of excess acetone and added IPA.
The yield is much better this time, surely because I took time to completly deplete the NH3 generator. Last try I must have wasted at least half the
NH4Cl this way, as alot of gas remains in the slurry. Next time, I might consider simply heating 20% ammonia to generate the gas, as I have no more
NH4Cl left.
the gradually addition of the acid during the reflux just didn't inspire me. I was scared of neutralizing too much unreacted NH3, and end up with a
bunch of ammonium sulfate. Considering that the H2SO4 didn't immediatly react with the atmospher inside the flask as it was introduced during the
titration, i think it was a good idea.
I added a extra portion of acetone before finishing the gassing, as the solution was getting saturated, and I didn't want to waste any NH3. Adding a
little more methanol could have helped.
Hopefully the product isn't too contaminated with ammonium sulfate, and a simple MeOH extraction and subsequent recrysatllization will be enough. A mp
determination is in order, though I have yet to find the litterature value for triacetoneamine hydrogenosulfate.
Conclusion: I would really recommend this recation to anyone desirable to obtaining a fair amount of TAA. The gassing stage isn't such as hassle, as
it it virtually takes care of itself, and the workup with H2SO4 (or ethanolic oxalic acid) is very easy and requires very little work. The gassing
time can be shortened, it just that I went too slow at first; I'm impressed at how well the mixture absorbs the ammonia, even with a flow of bubbles
close to 3-4 bubbles/sec, the glove didn't get blown up entirely until the end were the solution was saturated. It was pretty impressive to see the
massive bubbling of the solution from NH3 release once gassing was stopped.
Side note: I have tried the procedure without gaseous ammonia I posted a few post back, using NH4Cl, K2CO3, MgCO3 and CaO (I didn't have any MgO at
hand, and was unsure about using either MgCO3 or CaO, so just tossed both in) into a sealed bottle kept at ~40°C. In one day it has already turned
blood red, and the solids have turned from very fine powders to a spongeous mush. I will update with the results once workep up.
[Edited on 1-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
MeOH extraction of the above crude bisulfate was tedious, so it was decided to purify the TAA via it's hydrate.
The MeOH-wet powder was dissolved in 300mL warm water, resulting in a yellow suspension of a fine white/beige solid, which was vacuum filtered
(calcium sulfate) and washed with a little dH2O.
The yellow solution was basified with conc. NaOH until pH>13, at which point the solution became cloudy, and smelled of ammonia. It was placed in
the fridge, then in the freezer for 45min.
the mixture seemed to be entirely frozen. It was left to warm up for 15min, then vacuum filtered. It had turned to a gelatinous mass, which was long
to filter. Gelatinous scales slowly collected.


After testing their insolubility in acetone, the gel-like cake was flooded and triturated under 150mL acetone for 5min, vacuum disconnected, and
vacuum filtered again. Beautifull white scales were collected, pressed down and dried by suction for 30min, then layed down under a lamp to air dry.


After 4h, the total weight of pur 2,2,6,6-tetramethyl-4-oxopiperidine hydrate was : 62.0g.
EDIT: a second crop of the hydrate was obtained by adding K2CO3 to the filtrate and placing it in the freezer. The white crystals formed were vacuum
filtered, and washed with acetone before being dried by suction and under a lamp. The solid is much more "soggy" and is possibly contaminated by
K2CO3, although it melts around 50-60°C.
I'm unsure of which solvent to use to recrysatllize it. I think I recall undried ether been mentionned somewhere, but can't find the mention again.
What could I use, water? It might not be the most effective way to remove K2CO3..
Well, next step is NaBH4 reduction! I now have some TAA hydrate and recrystallized bisulfate (from the MeOH cristn) that will be properly
characterized.
BTW, can anyone suggest a easy and good yielding method of reducing oximes? I was thinking of doing a CTH to reduce the oxime of 4-oxo-TEMPO or TAA
(depending on reducer used, hydrogenation reduces the nitrosyl radical IIRC) as I've got some experience in the field, but if I could find a another
good reduction that doesn't involve Pd/C (or messy zinc), I would be happy. I have heard one unreferenced proposition of using NaBH4 alone, but
haven't found much info so I doubt it a bit. Any suggestions welcome.
Still the crude crop to purify and the unrecrystallized TAA.HSO4 to add.
[Edited on 3-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
guy
National Hazard
Posts: 982
Registered: 14-4-2004
Location: California, USA
Member Is Offline
Mood: Catalytic!
|
|
wow beautiful pure crystals!!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Thank you very much Guy
Apparently when purified via a salt, the TAA anhydrous freebase is pure enough to crisatllize at room temp, and forms beautfillu small white needle!
The problem is the mp is slightly depressed and in the day when it's hot they completly melt to a yellow oil, and crisatllize back at night
The hydrate stays as is though, and seems like the ideal way of storing TAA, no need of basifying for any further reactions, and if the nahydrous form
is needed, a reflux in toluene with a dean stark or similar should be just enough!
Gaseous ammonia is definatively the way to go!
EDIT: after some though, I'ev decided on posting all my TEMPO-related work in this thread, instead of making a thread for each compound, to regroupe
all the informations on theses derivatives in one big thread.
Synthesis of 4-hydroxy-2,2,6,6-tetramethylpiperidine by NaBH4 reduction of triacetoneamine
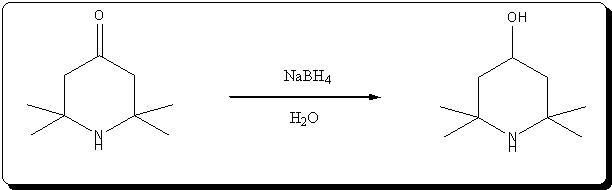
Ref:
Effect of electrostatic factors on the stereochemistry of the hydride reduction of ketones of the piperidine series
É. A. Mistryukov
Russian Chemical Bulletin (14)10; 1788-1792 (1965)
Very kindly provided by Solo in the ref forum.
Ina 100mL 3-neck RBF, equipped with a condenser, a addition funnel, a thermometer and magnetic stirring, 4.33g (25,00 mmol) of
2,2,6,6-tetramethyl-4-oxopiperidine mono-hydrate (preparation detailed a few posts above) were dissolved in 32ml of dH2O. the dissolution took a few
minutes and was endothermique. The solution was then cooled down to under 5°C in a ice bath.



A solution of 0.53g (14.03 mmol, 2.24 hydride equivalents) NaBH4 in 13mL dH2O containing a few mg of NaOH (to stabilize the hydride) was prepared (a
little fizzing at first, that quickly stopped) and charged in the addition funnel. With steady stirring, the hydride was added dropwise over 10min to
the cooled TAA solution. There was no noticeable appearance change, or visible bubbling. The addition funnel was rinsed with a few mL of MeOH, which
caused some TAA hydrate to crash out of solution. A little dH2O was added until complete dissolution. Once the addition was finished, the flask was
stirred in the ice bath for another 10min, then at RT (15°C) for 2h30.

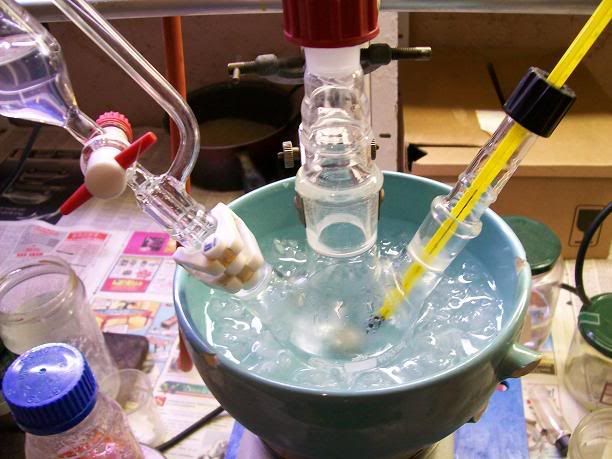

5mL of 31% HCl were then charged in the addition funnel, and added dropwise, evry slwoly at first as each drop caused massive H2 evolution. The flask
was immersed in a cold water bath, and the temperature kept under 20°C. After ~3mL of acid ahd been added, the gas evolution calmed down at each drop
then ceased completly. The rest of the acid was added in one portion. The completly colorless solution had a pH under 5 (universal pH paper), and had
a smell pretty similar to that of TAA freebase, but noticeably different.
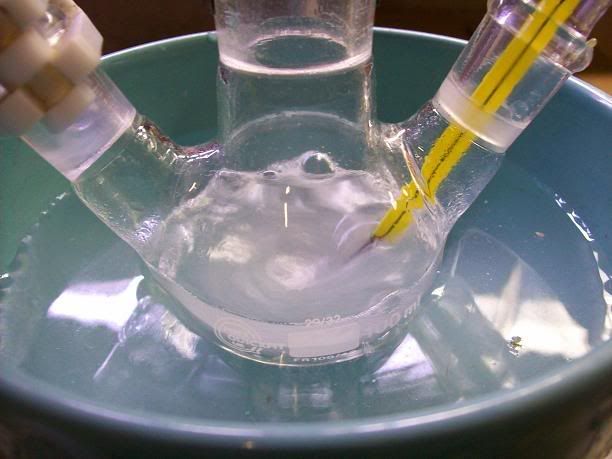

The solution was transfered in a 100mL single neck RBF, and place din the fridge awaiting vacuum distn of half the water tomorow.
Update will be posted when I get time to, possibly not until a day or two (that's why I preferred starting now, pretty busy at work at the moment).
The reaction looks very easy and manageable. Hopefully removal of most the water could be avoided by using more concentrated solutions (or adding
solid NaBH4) and saturating with NaCl or K2CO3, but for the first time I'm sticking to the article by the letter (well nearly ).
Direct oxidation to the nitrosyl in a one pot reaction could be possible, I don't think the borate salts can cause problems, actually perborate
formed by action of H2O2 on borax could be a good oxidant for this type of reaction!
It would just take neutralizing the excess hydride with hydrochloric or acetic acid (directly generating acetate co-catalyst in situ), adding
(bi)carbonate, and slowly adding H2O2 at 50°C.. Both reactions could be done pretty quickly, the reduction is surely finished in less than a hour
(TLC could confirm that), and the oxidation hardly taking more time than that. Place in the fridge, filter the next evening, recrysatllize et voilà!
Let's cross fingers.
[Edited on 4-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
detritus
Harmless
Posts: 19
Registered: 27-4-2008
Member Is Offline
Mood: No Mood
|
|
Wow. You are really the TEMPO master, Klute!
I just wanted to chime in that it seems gaseous NH3 is really not needed from what I'd found. I'm sure using gas would be better, but for those who
are too lazy to make a gas generator, fair results can be obtained by simply mixing the ammonium salt and a strong base under acetone.
I ran a few small-scale experiments comparing various ammonia salts and various bases and found that almost any ammonia salt and almost any base mixed
in situ with acetone will (after time) turn yellow, orange, then deep red. No heating was used, just time.
Lack of water seems to help the condensation but some water seems to help get a complete degradation of the ammonium salt. So hydrated salts like
ammonium acetate combined with your standard KOH or NaOH in an excess of acetone seem to work well, going bright yellow in just 1-2 days at room temp.
Color goes to orange after 4 days and is deep red after 8. I expect NH4Cl and NaOH would be even better, but I didn't make up any chloride.
I did not work any of these up as I was just looking for the speed of color change, but I did notice that with an excess of acetone, two layers easily
form if a little water is added. Maybe I'll run another test soon with the sulfate workup.
Pardon my bad form, please! This is just a intellectual exercise for me as I don't really have any use for TEMPO, so I only hope to contribute a
little to those who are interested but don't have all the fancy glass like Klute. As far as I can tell, this condensation is pretty easy - so more of
us should give it a try!
[Edited on 4-6-2008 by detritus]
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Very interesting! I am currently trying method using only ammonium chloride potassium carbonate and MgO or CaO, it's looking good (keeping it at 50°C
for afew days..).
Beware, color change doesn't necessairly imply triacetoneamine formation! At room temp the formation of acetonine and diacetoneamine are
predominant! Very long times (weeks) at RT or some heating is apparently needed to obtain good amoutns of triacetoneamine.
Indeed, the reaction seems to go well enough even with water, certain methods use NH4OH as ammonia source with (claimed) good yields). I'm sure
refluxing NH4OH with acetone and a catalyst would be a viable way of obtaining TAA, but you cna easily salt it out like with anhydrous conditions. At
least one tedious ddistn would be needed.
I agree using gaseous ammonia isn't very practical for most (even if it's much more simple that mmost thing, and relatively odorless with enouhg
precautions), but I consider this to be the best way I've tried until now. Doig it on a rather large scale doesn't imply that much extra work, and
then you are fitted with TAA for a loong time.. (keep in mind the oxidations are quite high yielding and the amount of TEMPO needed is rarely more
than 1-5% molar..)
I'd be delighteed if anyone tried out more preparations and isolated some TAA with much less effort!
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
| Quote: | Originally posted by detritus
I just wanted to chime in that it seems gaseous NH3 is really not needed from what I'd found. I'm sure using gas would be better, but for those who
are too lazy to make a gas generator, fair results can be obtained by simply mixing the ammonium salt and a strong base under acetone.
I ran a few small-scale experiments comparing various ammonia salts and various bases and found that almost any ammonia salt and almost any base mixed
in situ with acetone will (after time) turn yellow, orange, then deep red. No heating was used, just time.
|
Acetone will condense under the influence of bases to form progressively darker colored products as you describe even in the absence of ammonia. The
procedure you describe might work but I would be wary of making conclusions on the basis of color.
PGP Key and corresponding e-mail address
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Hum, bad surprise with the NaBH4 reduction!
2/3 of the water from the acidified solution was removed under vacuum, between 35 and 45°C (fluctuating because of irregular take off). the totally
colorless solution was then basified with K2CO3 until CO2 evolution stopped, and more carbonate added to acheive saturation. The solution turned
milky, and was extracted with 3x10mL DCM. A very fine white solid caused a thick emulsion as it stuck to the interface. The biphasic mixture was
filtered through a cotton plug to remove most of it, the collected solid was then extracted with 5mL DCM. The extracts were directly dried, and
solvent removed by simple distn, leaving.... nothing.
Nada, no a trace of solid or oil. A empty flask. That rarely happens! Confused, I though maybe the carbonate wasn't basic enough to freebase the
compound, although it is employed in the procedure I worked with, so I added some NaOH to the aqueous mixture. It turned milky, the distd DCM used to
extract the solution again, which turned clear, the solid (I mixed it with aqueous back again) filtered through a cotton plug, and the extract dried
and distd. TLC indicated a very eluted spot (AcOEt), so I considered that this time it had worked, and the NaOH was required to base the amine.
Upon removal of the solvent, there seemed to have a very small amount of oil left (could be mainly DCM, I didn't want to apply to much vacuum to
avoid eventual sublimation). The flask was cooled, and a little (1-2mL) pet ether added. There seemed to be a infinite amount of white solid appear,
but it could just be dust. After 30min in the fridge, still nothing (mp of the coumpound 127°C)
As a last ressort, I considered that the white solid that I considered to be borate salts from the hydride, was the product,a nd that it was
insoluble in DCM (unlikely). It didn't melt above 130°C, and had the appearance of an inorganic compound when heated on the hotplate. Insoluble in
water, DCM, or alcohol.
Were did my product go? Have I just discovered a reaction that is an exception to the law of conservation of matter? I'd prefer to get some
4-hydroxytetramethylpiperdine actually, more usefull to me right now than revoking the basics of chemistry
The only thing I can think of is that either:
-removal of the water in presence of acid, hence heating to 50°C for a long period ina cidic media, somehow degarded my product (condensations,
polymerisations?), giving a neutral water-soluble compound. Maybe it is more sensible than the compounds the authors of the article used (I doubt so:
3-methylpiperidone)
-the freebase is EXTREMELY soluble in water and not possible to extarct with DCM (doubt so, I would ahev at least extracted a minute amount).
-The TAA hydrate is not TAA hydrate (doubt so, mp is perfect). I will try a marquis test tomorow to confirm (shoudl react is it's not an amine,
no?), and possibly a hinsberg test with TsCl, should give an insoluble product upon action of a base.
-I have managed to make matter disappear in a vortex or a 9th dimension or something. In that case I wish to name this phenomenon "Klute's dilema
effect".
I will try the reaction again using methanol. I'm not sure how to dry the hydrate, heating the solid under vacuum before adding solvent? Distn with
dry IPA? Dean stark with a little toluene, then diluted with methanol?
I'm really surprised the reaction didn't work out, and really can't see where the problem comes from. The litterature made me confident this reaction
would be a breeze, I followed the procedure exactly...
Any suggetsions?
[Edited on 5-6-2008 by Klute]
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
One of my professors often used to say: "You ain't a real organic chemist until one day you manage to isolate sodium sulfate and believe it to be your
product!"
Sodium sulfate decahydrate: mp = 32.4 °C, insoluble in acetone
2,2,6,6-Tetramethyl-4-piperidone monohydrate: mp = 59 - 61 (°C)
So, the question is: Are you are real organic chemist or not?
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Hehe.. Well that aready happened to me in the past, so I'm already an organic chemist
I did a hinsberg test on my supposed TAA hydrate and a blank with Na2SO4. I think my TsCl was bad becasue in the two test a white precipitate formed
(much more with the TAA though), but even with the blank test it didn't dissolve in 10% NaOH, eevn with 20min heatinbg at 60°C..
I did a marquis test, the TAA dissolve dentirely inthe conc. H2SO4/formol, the Na2SO4 just formed a thick insoluble white block, with absolutely no
colour change (I used a tainted sulfuric acid though, so hard to see minor colour change).
I melted hydrated Na2SO4 and TAA hydrate one next to each other on the hotplate (cold at first), the Na2SO4 mellted a few minute sbefor ethe TAA, and
then formed an dry white solid (the anhydrous salt), were as the TAA melted and the quickly satrted to turn brown and fume.. I really doubt the TAA
isn't what it seems to be. But i would like to be able to determine is there are any inorganic salts present, especially in the second crop (there
surely, considering the change in apparance).
I purified the 4-oxo-TEMPO by column chromatography, beautifull blue substance came first, surely a unsaturated nitro- ring opening product described
in the article KMnO4 posted. I've kept the fraction to isolate it on rainy day to see if it's the crisatllin one or the oil. I guess removing the
ethyl acetate at atmospheric in presence of acetic acid caused some ring opening. Next time I will use DCM and wash the extract before removing
solvent.
Pictures to come in a few days; I will need to put a halt to reactions for a while , going to Paris for a few days.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
I used 5% AcOEt in toluene at the beggining, to elute most of the blue fraction, than gradually came up to 20% AcOEt in toluene to elute the
4-oxo-TEMPO: worked like a charm, and offered a very good seperation (there was a blank fration, no UV-visible compounds for ~50mL between the blue
and orange fraction, using 5uL samples on the plate (twice the amount usually used, most of the time hardly 0.5uL is needed to get a spot with 1%
solutions).
The solvent was removed under vacuum, to give a red oil that crisatllized once excess toluene had evaporated off at RT to give a low-melting red
solid. It will be recrysatllized with pet ether to try and get a sharper mp.
Pictures of the column:






First blue fraction:

it's surely one of the two unsaturated nitro ring-opening compounds mentionned in the article KMnO4 posted and out of which I used the procedure for
the oxydation. They claim ring-opening can happen on workup of the product, the ethyl acetate was removed at atm pressure and contained some acetic
acid. It seems that's all it takes. I will remove the solvent latter on and see which compoud it is (one is an oil, the other a crisatllin substance)
The orange fraction (pur by TLC) once solvent is removed under vacuum.

Picture of the pur product will arrive when done.
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Klute
International Hazard
Posts: 1378
Registered: 18-10-2006
Location: France
Member Is Offline
Mood: No Mood
|
|
Ok, I have tried another oxidation with the supposed TAA hydrate, and complete lack of isolated product has lead me to think the product is indeed a
mixture of sodium sulfate and potassium carbonate, along with a little amount of TAA hydrate, enough to give a faint amine odour and a colour change
with a marquis... God damn it!
I have trie dthe oxidation with the hydrosulfate I isolated, definitevly is the right compound as once basified it release this pale orange oil with
the caarcteristic TAA smell... So I have 50g of beautiufull hydrated Na2SO4 and no more TAA, great!
I will work up the TAA preparation with NH4Cl, K2CO3 and Mgo/CaO and see what gives, and might consider trying out a new synth, but without gaseous
ammonia. Refluxing NH4OH with acetone and CaCl2/NH4Cl overnight might be enough, IIRC Guy tried out a simialr thing. Guy, if you are reading this, how
did that run go? How did you isolate your product?
Details on the oxidation to come. I've decied on oxidizing evrything in one go, and thenr educing the nitrosyl to the hydroxy-TEMPO, the oxime and
the amine will wait for a new TAA batch...
\"You can battle with a demon, you can embrace a demon; what the hell can you do with a fucking spiritual computer?\"
-Alice Parr
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
I have yet to see a amine hydrogensulfate that is insoluble in alcohols, particularly methanol. And you recrystallized the crude product from
methanol, so perhaps that was the crucial error.
|
|
|
| Pages:
1
2
3
4
5 |
|










