| Pages:
1
2
3
4
5 |
Sandmeyer
National Hazard

Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
yO! Nope. And no radicalz either... Ho-Ho-Ho...
|
|
|
cheeseandbaloney
Harmless
Posts: 21
Registered: 4-4-2008
Member Is Offline
Mood: No Mood
|
|
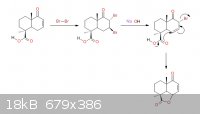
hmmmm so first hope your holidays went fantastically grand, second! Sandmeyer, goofed around with it for a bit tonight hope i'm on the right track:
|
|
|
Sandmeyer
National Hazard
Posts: 784
Registered: 9-1-2005
Location: Internet
Member Is Offline
Mood: abbastanza bene
|
|
Yeah, you've got it cheezy -- it's an Sn2' all-right... 
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Sorry, Nicodem, i dont know how all those thoughts were coming in my mind, but now i understand you are right. Arrow-drawn mechanism does not mean
polarization of any atoms in transition state, which i first believed to be, what ever direction this flow goes. As well, polarization in cases of
some hetero-DA reactions transition state does not mean there is no electron pairs flow or it is somewhat impaired. The problem of my ignorance should
have come from observation, that most hetero-DA's reaction mechanisms i seen were drawn as electron pair flow in only one direction, the same that is
taking place when reaction is not concerted. Then i decided that another direction of flow is less favorable, that electron pairs flow should include
a polarization in transition state, and that it may be not ideal and displaced by polar interations. Now i understand that direction of electron pairs
flow does not mean anything. Thanks. Pretty much loved your example about luna park.
[Edited on 1-2-2011 by Ebao-lu]
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Could someone explain a mechanism of this sodium sulfite reduction?
The book where it was found is here

[Edited on 1-2-2011 by Ebao-lu]
[Edited on 1-2-2011 by Ebao-lu]
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
Hi all,
at the risk of starting a new thread and looking like an idiot, I've decided to post my question here instead.
I am looking at a paper where the amidification of a carboxylic acid is done with POCl3;
R-COOH + POCl3 -> [intermediate] --- (R,R-NH) ---> R-C(O)N-R,R
At first glance I would assume one to use SOCl2, or oxalyl chloride to get to the acid chloride, and go by amidification from there. But instead POCl3
is used.
Under what circumstances would POCl3 be beneficial?
All I could find was that POCl3 is used extensively in biochemistry for some kind of coupling (sorry it escapes me) or for the dehydratioon of amides,
giving nitriles...
Having seen the paper used POCl3 I had assumed that possibly the starting material was not an acid, but an amide. The amide made into a nitrile, and
undergoes hydrolysis to make an acid, however this is not the case. The starting material IS and acid, and there is ONLY the addition of POCl3
and the corresponding amine to give an amide.
So - whats the deal with POCl3?
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by GreenD  | | The starting material IS and acid, and there is ONLY the addition of POCl3 and the corresponding amine to give an amide.
|
Perhaps it was some cheap amine that can be used in large excess. Otherwise it would be a bit of irrational to use POCl3 when it leaves a bunch of
amine consuming side products after the COOH to COCl transformation.
| Quote: | | So - whats the deal with POCl3? |
It leaves non-volatile side products, while oxalyl chloride and SOCl2 are more suitable, since all you need to do to get a crude acyl chloride is to
rotavap the reaction mixture (or distil via a distillation column in case of lower boiling acyl chlorides).
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
Does SOCl2 or oxalyl chloride react with secondary cyclic amines? Indoles?
Reactive towards anything other than alcohols and acids? Chemistry book says "Make sure you know the mechanism - thionyl chloride reacts in many
ways."
It acts as a lewis acid - and I believe an indolic amine is stable enough to go without side reaction.
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by GreenD | Does SOCl2 or oxalyl chloride react with secondary cyclic amines? Indoles?
Reactive towards anything other than alcohols and acids? Chemistry book says "Make sure you know the mechanism - thionyl chloride reacts in many
ways."
It acts as a lewis acid - and I believe an indolic amine is stable enough to go without side reaction. |
Indole gets quantitatively C-acylated by oxalyl chloride already at 0 C, requiring no catalyst. I don't know about its reaction with SOCl2, but I
would suspect it to react with it as well. However, indole is not a "secondary cyclic amine". Secondary amines, cyclic or not, vigorously react with
oxalyl chloride or SOCl2 in a different way. They get N-acylated or N-sulfinated respectively, to give oxalamides or sulfinamides.
In general, oxalyl chloride and SOCl2 can react with any nucleophilic enough functional group. Under acidic catalysis they even react with weak
pi-nucleophiles like benzene and alkenes.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
Ebao-lu:
This is an interesting reduction of tribromoimidazole - primarily because most of us don't often think about simple reagents like sodium sulfite.
That being said sodium sulfite is a fairly versatile reducing agent, and is dirt cheap. I don't know the mechanism for sure, but I feel bad that no
one's ventured a guess, so I will. This leaves unanswered as to why this reaction can achieve selective (or atleast somewhat selective)
di-debromination. Given the yield, it's quite possible that this outcome is controlled by optimizing reaction time rather than pure thermodynamics or
so.
What I've drawn is a combination of some literature precedent (see below). Brominated hydroxyphenols are easily debrominated with sodium sulfite,
whereas their monomethyl ether counterparts are not reduced under the same conditions. This is likely due to the fact that the hydroxyphenol undergoes
facile tautomerization to a keto or quinone form. For this reason, invoking addition of sulfite via a more reactive tautomer of imidazole seems
necessary. Given that a solution of sodium sulfite is only mildly basic, a proton catalyzed tautomerization seems OK.
It seems that either sulfite or water can act as the end reductant, yielding either the bromosulfate ion or hypobromite, respectively.
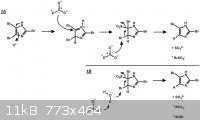
Refs:
1.) Arch. Biochem. and Biophys., (1974), 161(2), 632-637.
2.) J. Am. Chem. Soc., (1952), 74(6), 1601–1602
|
|
|
Ebao-lu
Unregistered
Posts: N/A
Registered: N/A
Member Is Offline
|
|
Yes, Arrhenius i think you are close to truth. That should be some kind of ionic substitution "at positive halogen". It is known to take place in some
chlorinations with CCl4(those that are not radical) and a-halosulfones reduction, SO2Cl2 chlorinations i suppose have similar mechanism
In this article it is even called "SNCl+" substitution, i don't know if it is widely accepted name, so here we should have SNBr+
http://www.sciencedirect.com/science/article/pii/S0040402001...
Thanks for information about brominated hydroxyphenols reduction with Na2SO3, that makes sence. Here we should have something similar.
[Edited on 26-8-2011 by Ebao-lu]
[Edited on 26-8-2011 by Ebao-lu]
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
How's that work?!!?
Hey all. I'm a bit saddened by the Organic stickies. It seems people are more interested in benzaldehyde and Duff reactions than mechanisms!!  Shocking! So I thought maybe we could work through some new problems. I'm a firm
believer that you should know *how* your synthesis is working, not just how to do it. Shocking! So I thought maybe we could work through some new problems. I'm a firm
believer that you should know *how* your synthesis is working, not just how to do it.
Here are a few rules for writing any mechanism:
- Don't break the rules.
- Don't skip steps.
- Identify a nucleophile and an electrophile and:
1.) make a bond
2.) break a bond
3.) draw the result
- Special case (e.g. pericyclic rxns) don't have a nucleophile, so this won't work
- Try to balance reaction equations. Organic chemists lose sight of this, but this basic concept is still important!
Fluorescein:
First synthesized in 1871. Basically, just mix resorcinol, phthalic anhydride, some sulfuric acid, and heat it a bit - someone should write up a
prep. The product is intensely fluorescent due to the electronic structure and extended conjugation in 2.
1.) Propose a mechanism for this reaction.
2.) For those unfamiliar, the 'delta' means heating. Why do you need to heat this reaction? What steps require this energy?
3.) Wikipedia calls this a "Friedel-Crafts" reaction. There's no AlCl3, FeCl3 or an acid chloride, so why is this still a Friedel-Crafts reaction?

In the Grignard reaction of ethylmagnesium bromide with benzaldehyde, the anticipated secondary alcohol 3 is isolated as the
major product, along with minor impurity 4. Indeed, this minor impurity is found in most reactions with organometal nucleophiles.
1.) Draw a mechanism for the formation of 4.
2.) What is the name of this reaction?
3.) What is the oxidation state of the aldehyde carbon in the starting benzaldehyde? and in the products? What is the oxidant or reducing agent that
promotes this redox, and what is its oxidation state throughout the reaction?

Magpie and others have worked on the synthesis of furfural, 6, from pentoses. The Org Synth prep prepares furfural from corncobs.
1.)What is the mechanism (following the rules!) for this reaction? (if you want to cheat, it's hidden in Magpie's thread).
Furfural can be used to prepare butenolide, 7.
2.) What is the mechanism for the reaction of 6 to 7?
3.) What is the name of this reaction?

[Edited on 5-12-2011 by Arrhenius]
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
I have a question related to a recent synthesis of mine. I was trying to work through the reaction mechanism and came to a sticking point. Please see
below. While I made phenylacetylene, from styrene, the common undergrad experiment utilizing trans-stilbene is a simpler system and the same mechanism
should apply. First, the alkene is brominated, giving the trans/anti addition product. This is heated with an alkali hydroxide in a high boiling
solvent (usually triethylene glycol) to induce a double dehydrohalogenation. I see this described usually as two E2 eliminations. If we do the first
E2, we find that the product is always E, which lacks an antiperiplanar H and Br for the second elimination.
I've heard that synperiplanar can E2 as well, but the transition state is significantly higher energy. Does the reaction proceed this way, does the
high temperature induce a double-bond isomerization, or does it go by a different mechanism alltogether? E1 would require spontaneous formation of a
vinylic carbocation, which I find exceedingly unlikely.
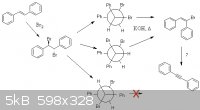
[Edited on 1-30-12 by UnintentionalChaos]
[Edited on 1-30-12 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
DJF90
International Hazard
Posts: 2266
Registered: 15-12-2007
Location: At the bench
Member Is Offline
Mood: No Mood
|
|
Double dehalogenation should only be possible when the product would be a terminal alkyne, as with styrene dibromide. The product you have drawn
(1,2-diphenylacetylene) would likely have to be prepared via a sonogashira coupling. Do you have a reference stating that the reaction you have drawn
is possible?
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
| Quote: Originally posted by DJF90 | | Double dehalogenation should only be possible when the product would be a terminal alkyne, as with styrene dibromide. The product you have drawn
(1,2-diphenylacetylene) would likely have to be prepared via a sonogashira coupling. Do you have a reference stating that the reaction you have drawn
is possible? |
http://www.orgsyn.org/orgsyn/orgsyn/prepContent.asp?prep=cv3...
Quite possible, in fact I did it as an undergraduate organic chem lab.
This question is actually about phenylacetylene, I figured that this system posed a similar problem. With styrene dibromide to phenylacetylene, the
two conformations for the first dehydrohalogenation (loss of the benzylic bromine) give a cis and a trans beta-bromostyrene. Yield of phenylacetylene
exceeds 50%
[Edited on 1-31-12 by UnintentionalChaos]
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
UnintentionalChaos
International Hazard
Posts: 1454
Registered: 9-12-2006
Location: Mars
Member Is Offline
Mood: Nucleophilic
|
|
I can't edit anymore. I found my answer in the lit. Looks like an E1cb.
http://144.206.159.178/FT/986/86265/1458858.pdf
Department of Redundancy Department - Now with paperwork!
'In organic synthesis, we call decomposition products "crap", however this is not a IUPAC approved nomenclature.' -Nicodem
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
I need this article, or could someone explain the mechanism within? Or both?
http://pubs.acs.org/doi/abs/10.1021/ja01469a048
ʃ Ψ*Ψ
Keepin' it real.
Check out my new collaborated site: MNMLimpact.com
|
|
|
zoombafu
Hazard to Others
Posts: 255
Registered: 21-11-2011
Location: U.S.
Member Is Offline
Mood: sciencey
|
|
I just got this book to go along with my organic chemistry textbook
<a
href="http://www.amazon.com/gp/product/1592577539/ref=as_li_ss_tl?ie=UTF8&tag=thehomche0e-20&linkCode=as2&camp=1789&creative=390957&am
p;creativeASIN=1592577539">The Complete Idiot's Guide to Organic Chemistry</a><img
src="http://www.assoc-amazon.com/e/ir?t=thehomche0e-20&l=as2&o=1&a=1592577539" width="1" height="1" border="0" alt="" style="border:none
!important; margin:0px !important;" />
It does a really good job of explaining mechanisms.
|
|
|
ScienceSquirrel
|
Thread Pruned
12-3-2012 at 14:26 |
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
GreenD:
The authors consider this a Beckmann rearrangement. Given that in this paper a variety of metals including nickel, copper and cobalt are demonstrated to affect the
isomerization of oximes to amides, the mechanism is likely Lewis acid activation - as opposed to redox. Bare in mind that Brønsted acids catalyze
this transformation as well. The mechanism in this case is as follows:

Oximes, in particular those derived from aldehydes (aldoximes), are configurationally stable. That is to say, the cis-/trans- stereochemistry about
the C=N bond does not readily interconvert at room temperature. This is important when we consider the mechanism, because only when the aldoxime
hydrogen and hydroxyl group are anti-periplanar can the E2 type elimination take place. I've drawn the orbitals involved to make this clear. The
electron pair in the C-H bond (large grey lobe) must populate the anti-bonding (small white lobe) of the breaking N-O bond, and symmetry requires they
be anti-periplanar. Nickel promotes this reaction by coordinating the oxygen, effectively weakening the N-O bond by pulling electron density away from
the oxygen center. You could also think of this as a resonance structure (shown) where there is a positive charge on oxygen and a bond to nickel.
From the resultant nitrile, the metal can coordinate nitrogen, making the carbon more electrophilic, and allow water to add. A proton transfer
(written ~H+) gets us to the amide.
[Edited on 29-3-2012 by Arrhenius]
|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
Arrhenius, bravo! That was great. The explanation of the anti-periplanar geometry, as well as the complexing. I was also stumped how the oxygen was
moving from the aldoxime to the amide, but it is actually from water. The stability of the aldoxime also cleared - thanks.
Here is numero 2. I've never encountered an alpha-alkelation before, and I have no idea where the hell to even start on this one.

ʃ Ψ*Ψ
Keepin' it real.
Check out my new collaborated site: MNMLimpact.com
|
|
|
Arrhenius
Hazard to Others
Posts: 282
Registered: 17-8-2008
Location: US & A
Member Is Offline
Mood: Stochastic
|
|
GreenD
It seems like you have some training in organic chemistry, but here are a few rules - mentioned previously - that I find extremely helpful for working
through 'unknown' mechanisms. Additionally, I've drawn reversible arrows where I believe reactions to be reversible. This is often important to
consider why reactions proceed to particular products. A majority of reactions involve a nucleophile and electrophile, and hence three rules
for drawing a mechanism to determine reaction producst/intermediates are:
1.) identify a nucleophile and an electrophile
2.) make a bond, break a bond (i.e. use arrows in a valence allowed manner)
3.) draw the result
Here's how to apply these to your second question. I've also drawn partial charges, where applicable, to indicate nucleophilic/electrophilic sites.
The key is not to get ahead of yourself, as this often leads to confusion if you try to skip a step (e.g. writing a proton transfer prematurely).
Step 1: The key realization to make here is that primary and secondary amines react with ketones and aldehydes in a somewhat
enthalpically favorable reaction, resulting in either an imine/enamine (tautomers) which are moderately stable, or an iminium which is strongly
electrophilic. Also, if an acid or base catalyst is present - in this case HCl - it will probably be utilized in the very first step of the mechanism;
in this case we protonate a ketone/formaldehyde.
Now, the next bit depends on how you run the reaction. I probably drew it differently than your stepwise reactions suggest. Step 1, strictly speaking,
probably forms the dimethyliminium ion of formaldehyde - this is much more electrophilic than formaldehyde itself. However, if the diethylketone were
present I believe it would proceed through an enamine catalyzed Mannich reaction as I've shown. Alternatively, the base in step 2 deprotonates the diethylketone, and the resulting enolate adds to
dimethylformaldiminium chloride. Enolates and enamines are 'isolelectronic', which is to say in both cases you can consider a resonance structure with
a negative charge on the carbon alpha to the carbonyl to suggest it is nucleophilic. While it's of little consequence here, the enamine will be of the
E (E=trans Z=cis) stereochemistry for thermodynamic reasons.
Step 3: Here, you must realize that amines react exhaustively with alkyl halides - when present in sufficient amounds - to form
tetraalkylammonium halides.
Step 4: The formal positive charge on nitrogen weakens the strength of the C-N bond to the neighboring CH2. Another way to say this
is that the NMe3+ becomes a better leaving group. Sodium hydroxide (pKa ~16) can deprotonate (only partially!) the ketone (pKa 20) to give the
thermodynamic enolate, which then gives rise to a type of Hoffmann elimination. This is somewhat reversible because the product possesses a Michael acceptor, but is enrtropically driven according to Le Chatelier's principle because trimethylamine is removed from the reaction as a gas.

|
|
|
GreenD
National Hazard
Posts: 623
Registered: 30-3-2011
Member Is Offline
Mood: Not really high anymore
|
|
Incredible!
Thank you so much. I will have to practice this a few times before I can "see" it if you know what I mean.
Quite an interesting reaction all together.
ʃ Ψ*Ψ
Keepin' it real.
Check out my new collaborated site: MNMLimpact.com
|
|
|
ZHANGNIUBI
Harmless
Posts: 28
Registered: 5-5-2011
Member Is Offline
Mood: No Mood
|
|
speak of the mechanisms, I have many interesting problems, I would say that are hard but useful when people want to learn total synthesis.
these problems, may have been known by some of us, called Fukuyama group meeting problems.
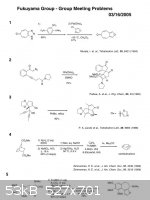
|
|
|
ZHANGNIUBI
Harmless
Posts: 28
Registered: 5-5-2011
Member Is Offline
Mood: No Mood
|
|
I'd say that whenever you know how Fukuyama's group meeting problem's mechanism, you ll be really fascinated.
|
|
|
2bfrank
Harmless
Posts: 5
Registered: 11-9-2009
Member Is Offline
Mood: No Mood
|
|
Im an IT dumbass, hence attached rather than post it directly. (been to busy to sort out how to do that, help would be good) but this mechanism was
associated with a synthetic and medicinal unit I am currently undertaking. Its predominantly using an iminium/enamine type catalyst with a Michaels
reaction and an Aldol reaction, I just thought it pretty cool. To me its poetic. Im still a little unclear, and taking my time with the stereo aspects
to it, but thought it a Pretty cool, albeit a simple mech.
Cheers
Attachment: ol100857s.pdf (439kB)
This file has been downloaded 1912 times
The most intelligent statements can be said so that an average person can understand, hence all the statements that require so much effort trying to
be understood, are an indicator that such a statement is not about presenting something of interest or intelligence, but rather an appearance of being
so.
|
|
|
| Pages:
1
2
3
4
5 |










