| Pages:
1
..
27
28
29
30
31
..
33 |
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
@nitro-genes
That is your work nitro-genes so no need to credit me as a co-author of your work, when all I did was help with some references and added some
untested ideas of my own. So you rightly own the copper[+1] cuprous / ascorbic acid reduction scheme. Let me own the magnesium idea... glycine ...ect.
especially if all that turns out to be a "seemed like a good idea" that doesn't work at all. I'm good at thinking ideas that work great on paper and
in theory but don't always work so great in the flask put at risk, when the laboratory and lab personnel are still around to report the interesting
results 
|
|
|
nitro-genes
International Hazard

Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
You were the first to recognize the product as picramic acid and provided references that pointed towards the yellow-greenish compound likely to be
copper picramate and also its solubility in HCl.
The magnesium and glycine idea may be worth pursueing, in the ideal scenario glycine complexation may keep the copper picramate soluble indeed and
allow the use of catalytic amounts of copper, without precipitating any magnesium picrate (like for sodium) during the reaction at near neutral or
slightly acidic pH.
As said before though, doing these reactions is very time and material consuming and there are many other experiments I would like to do with picric
acid, the copper/ascorbic reduction being mostly figured out.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Yeah I think because of the complex parallel reaction pathways that are possible that is a similar story with the Zinin reduction scheme, it is
definitely possible to tweak the reaction to obtain higher yields. Hennig was doing some sorting out of the variables for the Zinin reduction and got
the yields to go up from around 80% in the beginning to later in the 95% to 100% by what are really trivial changes to the basic process. Very
probably it is possible to refine the process if it follows the same for alternative schemes to the Zinin. These reduction schemes are somewhat
general and what works out good for one reduction may also work well for other reductions as is or with slight adjustments.
This is probably novel what you have reported for the ascorbic acid and copper sulfate used to produce picramic acid. So all the Google searches on
this topic will and already do lead here to SM. There is quite an extensive little compendium of obscure articles and experiments here at SM that is
nowhere else to be found.
I am thinking about the diazotization and the scheme you described using copper wire.
If the picramic acid is dissolved in nitric acid strong enough to form the picramic acid nitrate via the amphoteric property similarly as was observed
for the isopicramic acid, and copper wires are added, then the byproduct NO2 should work similarly as occurred when you were preparing the iso-DDNP or
p-DDNP by diazotizing isopicramic acid nitrate. By using a 50/50 mixture of the picramic acid and isopicramic acid it may be possible to produce a
racemic co-crystallized mixed isomer DDNP having unique properties and crystalline form if such a mixed isomer does exist and can be formed.
With regards to an alternative candidate transition metal salt that could function in small quantity as a regenerable catalyst not being sequestered
as picramate during the reduction any of the ones described by Girard in 1853 may work as well as does the copper for the actual reduction, but by
remaining in solution can be regenerated. For example, nickel sulfate, or possibly better nickel acetate or other organic acid nickel salt, is a
candidate.
And these reduction schemes may likewise be applicable to styphnic acid for reduction to styphnamic acid for diazotization to DDNR.


https://www.youtube.com/watch?v=R_ApBrVJD48 A Space Journey
jai guru deva om (Sanskrit mantra)
I give thanks to the heavenly teacher.
[Edited on 3/7/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Of all transition metals, copper seems to have the highest catalyzing effect on the reaction of ascorbic with oxidizers (see figure 5 in particular)
The Influence of Transition Metal Ions on the Kinetics
of Ascorbic Acid Oxidation by Methylene Blue in
Strongly Acidic Media
https://pdfs.semanticscholar.org/842b/11206b5630a7ba354d4727...
Iron comes in second, so I tried the reduction of picric to picramic using Fe(II)sulfate/ascorbic today:
Eperimental:
0.5 g of picric were added to a 20 ml beaker together with 15 ml water and brought to 60 C. The pH was adjusted to 7 using NaOH solution. Then 1.75
grams of ascorbic were measured out and added at once. About 200 mg of ferrous sulfate was added, which resulted in no color change or exotherm within
10 minutes. The pH was increased by dropwise additions of 5% household ammonia. Each added drop produced a dark black solution, which reverted to a
clear orange after few seconds. With each drop of ammonia, the solution did became a noticably darker shade of red in colour. After a pH of near
neutral was reached, the solution was a inmensely dark red in colour and gas formation became noticable. A sharp exotherm caused a rapid increase in
temperature to 80 C, with pronounced foaming. The end result was an incredibly dark red solution (light could hardly pass through it, even though the
solution was completely clear) Adding acetic acid to a pH of 3 precipitated some dark brown amorpous stuff.
So it seems iron can catalyze the reduction, though no picramic was produced at all. maybe the dark colour was from a nitro-diamino-phenol from over
reduction or the partial recution produces reacted with ascorbic or any of its decomposition products. Why copper(I) seems much more specific is
interesting.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I believe picramic acid is absolutely being produced even without the ascorbic acid to assist but conditions for the isolation and identification of
the product are not right. I'll get back with more on this later.
Over in the other thread I was reviewing this post
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
[Edited on 3/8/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
There is no way iron will work in high yield using ascorbic or using Fe(0). The Fe(II)(hydr)oxide method you are refering to produces only very small
amounts of picramic and very dark red solutions, likely indication aspecific reduction, as was also briefly mentioned in the french article you posted
a while back. None of these methods mention yield, except for the Fe(0)/NaCl/water method posted by Axt, which claimed something like 98% yield, I
don't buy it...Noone would be using sulfide reductions if this was the case.
From my experiment, sodium picrate in the presence of ascorbic acid and Fe(II) also does not result in any reduction, which is not surprising since it
was already knwon that boiling ferrous sulfate with sodium picrate does not result in any reduction. It seems, adding ammonia dropwise to the
picrate/ascorbic/Fe(II) solution was a best guess possible way to use ascorbic acid with fe(II) catalysis and it didn't work. No picramic was formed
and I don't see it working otherwise. Interesting why the copper works so well. It would be an interesting test to see whether some soluble cuprous
salt in a weak acid (something like cuprous acetate/acetic acid) would be able to reduce sodium picrate...
[Edited on 8-3-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
That article posted by Axt was published as a contribution from an American laboratory at the University of Chicago in what was the premier chemistry
professional journal of the era. It was in that same Chicago laboratory was built the world's first nuclear reactor.
The level of peer review for that article published 90+ years ago would very likely have been extreme and exhaustively thorough.
Berichte der deutschen chemischen Gesellschaft
http://onlinelibrary.wiley.com/wol1/doi/10.1002/cber.1927060...
Robert Edward Lyons, Ph.D. had been a Ph.D. and head of the university chemistry department for more than 30 years when that article was published.
http://webapp1.dlib.indiana.edu/archivesphotos/results/item....
In this post nearly four years ago I had done more study about the ferrous sulfate reduction
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
You are encountering conflict with lowering the pH for using ascorbic acid instead of an ascorbate salt and I think also the order or manner of
addition or both could be a factor.
I'll think more about the process and what may help.
"dark red solutions" standing in the cold sometimes produce some crystals of a picramate salt in good time 
Your "dark brown amorphous stuff" is likely picramic acid but the color is off due to the state of subdivision and probable impurity of iron.
If you check the amorphous stuff will probably redissolve in HCl to form the picramic acid hydrochloride and on dilution will likely precipitate
something looking more familiar.
I think you are making some overly broad general conclusions that aren't necessarily governing because of a very specific scenario where you see the
unexpected occur. Hang in there and sort it out.
Process chemistry is highly nuanced and what may seem to be insignificant small changes can produce a drastic change in the course of a reaction and
the resultant yield. There are an assortment of reactions where a small change in temperature or pH or the sequence and rate of addition or the form
in which the reactants are brought into contact and intermixed, solubilities and concentrations also can all have enormous bearing ....and this
reduction is definitely one of those more "fickle" reactions, evident from reading the earliest articles and later articles too. So, what on first
glance appears at first may not work, may later work even splendidly well when the precise conditions needed are determined and provided. Calibrating
the process conditions involves applied algebra that can't really be derived from one experiment. When you describe a dark red to almost black
solution along with the exotherm, that is a good indicator of reduction to sodium picramate which is a very strong dye and would remain dissolved in
super saturated solution in the hot alkaline liquid, but should crystallize out in the cold and on standing.
Something that could be occurring that makes the copper attractive and useful relates more to the insolubility of the copper picramate than to any
catalytic activity with ascorbic acid. The reduction may be enhanced by being solubility driven due to the product precipitating as fast as it forms.
That theory would be proven by use of other reducing agents than ascorbic acid in the presence of copper as a "solubility reagent" where basically the
product is appearing like a low solubility "spot test" result. The "complex" with copper may have nothing to do with ascorbic acid, but could be a
mixed valence copper salt, a double salt of cuprous picramate / cupric picramate. Only analysis of the intermediate precipitate would tell more or
tell conclusively what is occurring. With simultaneous multiple different reaction paths possible, there could be an indefinite mixture of products
of sodium picramate, cupric picramate, and cuprous picramate ....all present as possible mixed "picramate values" and likewise for other reductions
using other metal salts as reagents or catalysts.
[Edited on 3/8/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
On heating picric to its ignition point, there is a definite sign of reduction going on, maybe picramic can be isolated from the condensed vapours.
Granted, you have a point about my conclusion about the iron reduction never to reach high yield being premature. Noticed in the article they used
iron from which all organic contaminants were removed by glowing out (85% iron) maybe the oxydes introduced could be a factor. Still, I have a really
hard time believing the 100% yield mentioned, hardly any information on isolation and compound identification is present, except mention of sodium
carbonate extraction. As an amateur this may be acceptable to a certain extent, especially when on closer look, no definite conclusions are generally
made at all.
The mixed valence picramate theory is interesting, but with such an excess of ascorbic acid and the very fast conversion of Cu(II) to Cu(I) (Even in
the cold) I find it very hard to believe as well. A sodium double salt might also be possible, that is why I wrote "perhaps" in the report IIRC.
Looking at the properties of Cu(I) salts and there ease of hydrolysis in general, it seems strange so little copper(I)oxide is isolated under these
weakly acid conditions. Maybe most of the copper(I) remains as a Cu(I)ascorbyl radical complex during the reaction?
You mentioned at least 4 reactions to be possible during the copper/ascorbic reduction of picric, could you shed some more light on your thinking on
this matter?
[Edited on 9-3-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Quote: Originally posted by nitro-genes  | | On heating picric to its ignition point, there is a definite sign of reduction going on, maybe picramic can be isolated from the condensed vapours.
|
One ferrous picrate reduces another ferrous picrate to ferrous picramate just like one hand washing another. Didn't you get the memo that went out
about 90 years ago?  Sheesh...My dear missus, do hand me a new spoon feeding
spoon since this one I have been using is worn down to the nub. And fetch for me my German - English dictionary and reading glasses. Seriously the
end product is probably ferric picramate since likely the ferrous picramate is active as a reducing salt also, and there is a 6H+ represented by the
3:1 molar ratio of elemental Fe(++) to picric acid described by Lyons and Smith. Sheesh...My dear missus, do hand me a new spoon feeding
spoon since this one I have been using is worn down to the nub. And fetch for me my German - English dictionary and reading glasses. Seriously the
end product is probably ferric picramate since likely the ferrous picramate is active as a reducing salt also, and there is a 6H+ represented by the
3:1 molar ratio of elemental Fe(++) to picric acid described by Lyons and Smith.
| Quote: |
Granted, you have a point about my conclusion about the iron reduction never to reach high yield being premature. Noticed in the article they used
iron from which all organic contaminants were removed by glowing out (85% iron) maybe the oxydes introduced could be a factor. Still, I have a really
hard time believing the 100% yield mentioned, hardly any information on isolation and compound identification is present, except mention of sodium
carbonate extraction. As an amateur this may be acceptable to a certain extent, especially when on closer look, no definite conclusions are generally
made at all. |
You have me at a disadvantage because my German is awful.
Sponge iron or water jet cut powder iron would probably do better than 80 mesh iron filings.
This form of iron is what I was thinking should work well.
https://www.ebay.com/itm/Hoeganaes-Ancor-MH-100-Sponge-Iron-...
I still think zinc or aluminum amalgam would work fine. Aluminum amalgam or zinc amalgam is piss easy.
| Quote: |
The mixed valence picramate theory is interesting, but with such an excess of ascorbic acid and the very fast conversion of Cu(II) to Cu(I) (Even in
the cold) I find it very hard to believe as well. A sodium double salt might also be possible, that is why I wrote "perhaps" in the report IIRC.
Looking at the properties of Cu(I) salts and there ease of hydrolysis in general, it seems strange so little copper(I)oxide is isolated under these
weakly acid conditions. Maybe most of the copper(I) remains as a Cu(I)ascorbyl radical complex during the reaction? |
The ascorbyl or dehydroascorbyl could have a chelating effect but I doubt it is the predominating influence on the copper.
When future experiments are done if you are sticking with sodium as the alkali maybe half-neutralize the ascorbic acid solution to a 50/50 molar
ascorbic / sodium ascorbate. But magnesium would work better Reference the
Clayton article and discussion about pH control that Hennig and I were sorting out with regards to the different reduction yields being pH sensitive,
(in the other thread).
Picramate is a devil about pH sensitivity affecting yields, also temperature and reaction mixture concentration are other tricksters.
Regarding the possible formation of a mixed valence copper picramate double salt:
There is cupric picrate present in solution before the ascorbic acid is added and it is possible for that to be reduced to cupric picramate ...if
there is not a specifically selective reduction of the cupric to cuprous in preference to reduction of the picrate to picramate.
Of course if the reduction went to completion all that would be present in the end would be entirely cuprous picramate. However, if there was formed a
mixture of partly reduced and fully reduced copper picramates that tended to form an insoluble double salt there could be a mixed valence double
copper picramate that accounts for the insoluble product chemical composition showing a discrepancy on analysis from what should be the mole weight
and analysis for cuprous picramate.
It is uncertain what is the selectivity for the species being preferentially or first acted upon by the ascorbic acid .......is it the copper being
first reduced, or is it the picrate, or is it a mixture of both reactions? You see the uncertainty present in a chaotic system allows for several
different reductions and subsequent reactions for the reduced copper in particular, since its oxidation state can change. Reduced copper associated
with either picrate or picramate can operate as a reducing agent itself towards unreduced picrate.
| Quote: |
You mentioned at least 4 reactions to be possible during the copper/ascorbic reduction of picric, could you shed some more light on your thinking on
this matter?
[Edited on 9-3-2018 by nitro-genes] |
There is possibly a couple more that are pH and hydrolysis dependent
Reduction by ascorbic H++ (free ascorbic at < 4 pH)
Reduction by mono-dehydroascorbic H+
(cuprous ascorbate or sodium ascorbate)
Reduction by cuprous picrate (reduction by the Cu+1)
Reduction by cuprous picramate (reduction by the Cu +1)
Ascorbate will by itself reduce Picrate to Picramate
https://www.jstage.jst.go.jp/article/bcsj/65/4/65_4_1101/_ar...
Put on your thinking cap, and think about the concept of a regenerable catalyst needing to remain in solution, because copper won't do it. Obviously
copper is useful for isolation because of the insoluble precipitate it does form. But that same usefulness works counter to the concept of a
regenerable catalyst, because once the copper combines with the product it is sequestered by precipitation and leaves the reduction reaction, like a
player in a sports game that has scored and as a reward has been benched....that copper is no longer participating in the reduction reaction and has
been consumed and locked away in the insoluble precipitate. So then for a regenerable catalyst, a better candidate would be a metal that easily
changes oxidation states either direction, but behaves differently and does not form an insoluble precipitate with either the picric or picramic value, but remains in solution to be recycled and regenerated as an intermediate reducing
agent so that more usefulness as a catalyst is realized. Of course there would need also to be no precipitation of the catalyst due to reaction with
the Ascorbic Acid or Ascorbate Salt, and no other reduction reaction parameter such as pH that would provoke precipitation of the soluble catalyst.
So what transition metal salts would be likely catalyst candidates?
Manganese, iron, cobalt, and nickel are likely and each would probably be optimal at a particular pH, temperature, and concentration which would need
to be "process engineered" by the chemist / technician /operator keeping that pH and other parameters in mind with regards to order and time of
addition and the reagents used.
My intuition about these was first on Manganese due to good solubility of the picrate and picramate, but what is the sensitivity with Ascorbic or
Ascorbate is unknown. Nickel was another thought because of similar chemistry with copper but no report of an insoluble picramate to complicate
things.
My collateral thoughts about buffering using Magnesium as the base was likewise good solubility for the picrate and picramate and having a limited
swing for pH inherent by use of Magnesium for the base. Likewise using organic acid salts instead of mineral acid salts would dampen pH swings. And my
thoughts regarding glycine as potentially useful related to a twofold potential usefulness as an organic acid buffer and solubility enhancer via
chelation as well as a solubility enhancer for picric acid in a low pH reduction system which would enhance the activity of Ascorbic acid at a lower
and optimum pH of about 2-3 pH where free Ascorbic acid is at peak activity as a 2H (++) value reducing agent.

As a further thought, some reactions absolutely do run differently on infusion of reactant streams by a Sigma pump flow through a capillary into a
rapidly stirred reaction mixture as a finessed very gradual addition, rather than addition by "bucketful" splashes of drips for addition, or where
the reactants are simply dumped together "in a lump" and a mass reaction follows where the process on a molecular level is like a bar room brawl
It depends on the target material being made what addition scheme may work better and there can be quite a difference in yield for some of the
reactions, and even some reactions that can only be done by infusion to produce good results. I don't think this reduction is all that sensitive, but
some of the diazotizations for example like for nitrotetrazole are indeed that sensitive.
When you devised that diazotization scheme using copper wire as a source for NO2 gas evolution that essentially emulated a Sigma pumped infusion
diazotization scheme in situ, and is a lot simpler and more economical than using a Sigma infusion pump to very slowly inject a micro stream of
nitrite solution into the reaction mixture. That thinking cap is the one I was referencing.
[Edited on 3/10/2018 by Rosco Bodine]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
| Quote: Originally posted by Rosco Bodine | Diazodinitrophenol - as a free running crystalline powder from a nitric acid diazotisation
Abstract:
Diazoltisation of picramic acid to produce the explosive is effected in nitric acid in the absence of hydrochloric or sulphuric acids or their salts
and pref with as low a sodium content as possible. Conventional products tend to adhere to surfaces.
A translation of the attached patent FR2106904 may be useful
|
Attachment: Machine Translation English FR2106904A5 Diazodinitrophenol.pdf (18kB)
This file has been downloaded 934 times
Calcium nitrite could be an alternative means of diazotization to eliminate the sodium ion, if the calcium ion is not responsible also for lowering
the quality of crystals of DDNP similarly as the sodium ion. It would be easier to lower the concentration of calcium ion in a residual solution of
nitrous acid by use of an acid that would precipitate most of the calcium as an insoluble byproduct, sulfuric, phosphoric, oxalic, ect.
Calcium nitrite would also be easily converted to other nitrites by double decomposition reactions with carbonates or sulfates, ect. that would
precipitate the insoluble calcium salt and leave a solution of the desired different nitrite product.
An additional scheme for diazotization of picramic acid might employ an organic nitrite such as ethylene glycol nitrite or perhaps glycerol nitrite
used as the nitrosation reagent.
A gaseous nitrosation reagent such as methyl nitrite introduced through a dispersion tube into a picramic acid solution may also be workable.
Attachment: US4294813 Calcium Nitrite from Sodium Nitrite.pdf (103kB)
This file has been downloaded 738 times
Attachment: blinded.mid (27kB)
This file has been downloaded 1116 times
[Edited on 3/15/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Regarding the Cu(II)/ascorbic reduction of picric to picramic:
Would nano-copper itself be able to reduce picric? Or would this be a side reaction causing yield reduction instead. Noticed some mention of aminoacid
additions to control for smaller copper particle sizes from chemical reduction methods, is this what you had in mind with the glycine additions Rosco?
Not sure if the nano copper would be able to react easily again with copper(II)sulfate to form transient copper(I)sulfate. Would chloride content also
be a factor for this? From the second article attached it seems this might be the case, even in abscence of HCl.
Tried making some nano-copper from ascorbic reduction of copper(II)sulfate in water and boiling for an hour a few days ago. I somewhat doubt this
produces actual nano copper though, as the particle size appears larger instead (difficult to say though due to potential agglomerations formed). The
attached artcle mentions nano copper is produced best from dilute solutions at close to neutral pH and temperatures between 60-80 C. They also mention
the ascorbic to acts as a capping agent to keep the particles in suspension, though no supporting evidence for this is presented in the entire paper,
which would have been interesting to read. This might explain however the low 50% yield of copper powder from Cu(II) reduction by ascorbic, even in
the presence of a large excess of ascorbic. Only the larger copper particles (or agglomerates of nano-powder) are filtered off, leaving a lot of the
nano-copper in suspension.
Attachment: Experimental Investigation on the synthesis of copper nanoparticles by chemical reduction method - Copy.pdf (1.1MB)
This file has been downloaded 969 times
[Edited on 21-3-2018 by nitro-genes]
Attachment: Representation of the Solubility of CuCl in Solutions of Various Aqueous Chlorides - Copy.pdf (725kB)
This file has been downloaded 837 times
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Colloidal copper might be active as a reducer ....I'm not sure. Glycine would probably be helpful there as a sort of "emulsifier" or stabilizer that
would operate as a solubilizer / chelating agent ....which essentially "associates" metal and organic acid as a sort of loose covalent / quasi ionic
"salt" but not exactly a stable "electrical" salt like a usual mineral salt would be. The charge there for the compound association is minimal to
almost non-existent. A chelate is almost like a soap or detergent "wetting agent" like a very minimal "glue" on a "sticky note" attachment of two
things.
The "male" metal and "female" organic acid of a chelate are not joined in a tightly bonded "super glued" attachment, but are associated like "friends
with benefits" not involved in some deeply committed soul mate sort of bonded
relationship.
The organic acid offers mister macho metal a little joyous cathode stimulation / R&R / rejuvenation for the afternoon or as long as it can last
...until assertive competition arrives and it is time for girlfriend to go
My idea for using the glycine is raising the upper limit pH and soluble metal concentration where a metal hydroxide would precipitate, making the
reaction less sensitive to alkalinity. It was also a means of working with an organic acid salt of the metal in a system that would have inherent
buffering not present to as great an extent with mineral acid salts. Glycine would help tame the pH swings and the pH gradient in the reaction
mixture where additions are being made dropwise also, because each added drop has a local reaction condition that fades across a gradient until the
added drop of reactant becomes mixed thoroughly well with the bulk of the material being stirred in a beaker.
Adding a reactant solution dropwise is like dripping a red paint into a stirred bucket of white paint, and across the swirl of mixing different
materials is a million different shades of pink before long stirring makes the entire bucketful an even color pink. In a chemical reaction a buffer
helps narrow the range of extremes of pH that can occur only across a smaller limited gradient of pH for the mixing solutions.
The use of glycine in another scheme was as a solubility enhancer for picric acid that might be reducible to picramic acid or a picramate,
particularly the magnesium salt. I tried to give a detailed and coherent description earlier what were my thoughts on that. My focus and note on
magnesium having potential usefulness is explained more in this linked post in another thread.
http://www.sciencemadness.org/talk/viewthread.php?tid=433&am...
Note to moderator:
There needs to be an editing / export / merge done for the DDNP and picramic acid topic specific posts that have mislocated the discussion in another
thread making it impossible to keep track of the discussion.
See the enumerated 23 posts below need to be merged following this linked post in this thread
(1-12-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=439&am...
That merge should fill the chronological gap in this thread where the off topic posts are located in a different general topic.
Nitro-genes started this detour off topic by not recognizing the subject matter for the unknown compound he had made. So this (linked below) first
post "detour" off topic should be merged into this thread and all the posts listed should be exported to this topic thread, DDNP & related
compounds: The uber thread!
[1] (1-14-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[2] (1-15-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[3] (1-15-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[4] (1-15-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[5] (1-16-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[6] (1-16-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[7] (1-16-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[8] (1-17-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[9] (1-17-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[10] (1-17-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[11] (1-17-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[12] (1-17-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[13] (1-17-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[14] (1-23-2018) nitro-genes)
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[15] (1-23-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[16] (1-23-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[17] (1-24-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[18] (1-24-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[19] (1-24-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[20] (1-26-2018) nitro-genes
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[21] (1-26-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[22] (1-27-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
[23] (1-30-2018) Rosco Bodine
http://www.sciencemadness.org/talk/viewthread.php?tid=26572&...
Additional note to moderator:
There may need to later created a 3 branched bracketed grouping of this topic thread with the picramic acid from picric topic thread and the Picric
acid: different instructions topic thread since the three topics are an inherently related "trifecta" sort of overlapping discussion. That would help
organize the three related topics.
[Edited on 3/22/2018 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Would be interesting to know how exactly the reduction is occuring. Would something like this (Copper
phthalocyanine-3,4′,4″,4″′-tetrasulfonic acid) be "glueye" enough to allow for catalysis? Or would the ascorbic still reduce the copper right
back to the metal? It would be somewhat sterically hindered though.
https://www.chemicalbook.com/ProductChemicalPropertiesCB6113...
[Edited on 26-3-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Styphnamic acid!!! :
The copper/ascorbic method of reducing picric acid to picramic acid seemed surprisingly specific in reducing one ortho nitro group (the method was
posted on page 28 of this thread), even approaching the efficiency of the well known (hydro)sulfide reduction. I was curious whether the
copper/ascorbic reduction would produce similar results for styphnic as for picric, as also mentioned by Rosco Bodine a while back. The product upon
reduction of styphnic would be styphnamic acid, a pretty elusive compound, which is, (similar to it's diazo derivatives) only few times mentioned in
literature. The diazo derivatives itself are of little interest as energetic materials in practice due to incompatibility/stability issues and the
reported extreme friction sensitivities of their salts. The chemistry of both these selective reductions and diazo derivatives is still very
interesting IMO.
Basically the same approach for the reduction of styphnic was used as that for picric posted earlier. The styphnic was produced by hydrolysis of
3-aminopicric using 3 molar eqvts of NaOH and boiling the solution until no more smell of ammonia could be noticed (~30 min) followed by acidification
using HCl. Overall, the reduction of styphnic seems to behave very similar as that of picric. Curiously, whereas the reduction of picric produced
very dark brown solutions and produced a lot of gas during the reaction, the reduction of styphnic was without any noticable gas formation and no dark
brown solution was observed near the end of the reduction. Yield needs to be measured, though might be at least as good or even better as that for
picric.
Experimental:
Reduction of styphnic to styphnamic acid:
0.5 g of styphnic was added to a 100 ml beaker and water was added to 75 ml in total. Then, 0.27 g basic copper carbonate was added and the solution
heated until all styphnic had dissolved into a dark yellow solution and no more CO2 was produced. A bit of undissolved copper carbonate remained
because of the slight excess used. The copper styphnate solution was cooled to about 10 deg C. Next, 1.5 g ascorbic acid was weighed out and added at
once. After the ascorbic had dissolved, I let the reaction stirr in the cold for another 10 minutes, though no clouding or precipitation was observed.
The hotplate was turned on and the solution was heated slowly. When at 25 deg C. Slight clouding could be seen forming first, followed quickly by a
light green precipitate (very similar to that observed for the picrate reduction). Heating was continued and slowly brought up to about 65 deg. C. The
light green precipitate started to colour increasingly more yellow-greenish-brownish (also similar to the picrate reaction). After 30 minutes at 60
deg. C., the mixture was cooled down to room temperature and the precipitate left to settle. Most of the supernatant was decanted, then another 75 ml
of cold water was added and most decanted/siphoned off again. The beaker was added to the hotplate again, 10 ml water was added and warmed slightly
while dropwise additions of concentrated HCl were started. The greenish-yellow precipitate slowly turned a more orange colour and dark copper-reddish
crystals of styphnamic acid started forming. The solution was cooled down in the fridge, filtered and washed with water to remove all of the copper
and finally dried. The crystals of styphnamic acid look very similar to picramic, though with a more golden-brown note to it (See attached photo, some
cuprous styphnate seems to still be present). A rough melting point was taken on the hotplate and had a much higher melting point as picramic acid at
around 210-220 deg. C. (with extensive bubbling and decomposition).
Diazotization to dinitro diazoresorcinol or DDNR:
About 100-200 mg of the stypnamic acid was added to a 10 ml beaker together with 5 ml of 10% HCl. Some of the styphnamic acid dissolved, most remained
as suspension. This was cooled to 0 deg C. in an icebath and 2 ml's of water with a spatule of sodium nitrite dissolved was added dropwise. Here is
where it gets weird... Each drop of the nitrite solution produced a pretty dark
colour (maybe due to residual copper?, or an N-nitroso intermediate?). After only a few drops of the nitrite solution were added, all of the
styphnamic acid dissolved into a clear dark yellow-brownish solution. After stirring for another couple of minutes, a very light yellow precipitate
started to from (chloride salt of DDNR?). Thinking it was the DDNR itself, I reasoned adding another 5 ml of cold water would probably precipitate
more of the DDNR. To my surprise however all product dissolved again. Since there hadn't been any gas production during the diazotization itself, it
seemed unlikely the compound had decomposed somehow, as could have been the case for styphnamic containing an amino group in 2-position. To
precipitate any possible DDNR as the salt, solid sodium bicarbonate was added in small increments. When most of the HCl was neutralized, large amounts
of fine dark yellow needles started to precipitate, presumably the sodium salt of some DDNR isomer. Each drop of the nitrite solution produced a pretty dark
colour (maybe due to residual copper?, or an N-nitroso intermediate?). After only a few drops of the nitrite solution were added, all of the
styphnamic acid dissolved into a clear dark yellow-brownish solution. After stirring for another couple of minutes, a very light yellow precipitate
started to from (chloride salt of DDNR?). Thinking it was the DDNR itself, I reasoned adding another 5 ml of cold water would probably precipitate
more of the DDNR. To my surprise however all product dissolved again. Since there hadn't been any gas production during the diazotization itself, it
seemed unlikely the compound had decomposed somehow, as could have been the case for styphnamic containing an amino group in 2-position. To
precipitate any possible DDNR as the salt, solid sodium bicarbonate was added in small increments. When most of the HCl was neutralized, large amounts
of fine dark yellow needles started to precipitate, presumably the sodium salt of some DDNR isomer.
Since these salts are extremely dangerous (and I had made quite too much ) I
saved a few mg's and dissolved the rest in warm water again. When dry it behaves very energetic though, detonating in tiny amounts, much resembling
SADS.

What really puzzles me is how the presumed internal diazonium salt of styphnamic acid (DDNR) can actually seem more soluble in cold HCl as the
styphnamic acid itself...Also curious which isomer is formed during the reduction, maybe that could explain things....hmmm....It also doesn't seem
like the same compound as was obtained from the nitration of iso-picramic acid....pfff, I was expecting to close the loop and connect all dots here,
and then this?!?!?! ...if anyone has some thoughts on this, please share
them!!!
[Edited on 25-11-2018 by nitro-genes]
|
|
|
dave321
Harmless
Posts: 45
Registered: 22-11-2012
Member Is Offline
Mood: No Mood
|
|
i think there is a patent by josef kohler re the conversion of styphnic acid to styphnamic acid using tin chloride.
this was the diazotised and the strontium salt formed by further reaction.
you could try making the strontium salt , which i believe is quite a potent primary
i am sure i posted the paper in the ddnp thread somewhere previously
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Redoing the copper/ascorbic reduction of styphnic to styphnamic, this time using 1.5 g of styphnic, I was able to measure yield.
Running the reduction again....1.5 g of styphnic resulted in 1.20 g of styphnamic acid. Assuming styphnamic does not form any hydrates (which didn't
seem so when taking the melting point on the hotplate) this would translate to a 91% yield. Considering the scale of the reaction, any possible
remaining styphnamic in the dilute HCl filtrate, and the fact I was too lazy to really scrape all styphnamic acid of the filter paper, I think the
reduction might be almost quantitative... amazing!!! 
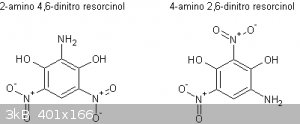
Patent US4246052 deals with the styphnic reduction indeed, unfortunately yields of styphnamic acid are not mentioned, only that a 63% yield of an
unspecified diazo derivative after diazotization is realized. How to interpret this depends on whether both the 2 and 4-amino isomers (see attachment)
of styphnamic are able to form diazo compounds and whether SnCl2 acts as a specific reducing agent (don't think so). What isomers are formed during
the copper/ascorbic reduction I'm not sure. Assuming the copper/ascorbic reduction is facilitated by forming some transient complex associated with
any of the 2 hydroxylgroups of stypnamic, one would expect a 50:50 ration of 2 and 4-amino derivatives of styphnamic maybe?
Anyway, can't wait to experiment a bit with different diazotization schemes! The strontium salt might be interesting, though I don't have any
strontium salts available.
[Edited on 26-11-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Ok, I've run the diazotization of 200 mg styphnamic acid again, this time using concentrated (30%) hydrochloric acid and more according to the
conditions outlined in US4246052 (SnCl2 reduction of styphnic). The stypnamic acid was all dissolved in about 2 ml of warm hydrochloric and the
nitrite slowly added as concentrated solution at 0 deg. C. This time, small light yellow cubic crystals (chloride salt or free diazoquinone?)
separated after 15 minutes. After filtering off these crystals, adding the least amount of water to wet them, and letting this react with bicarbonate
until a pH of 6 was reached, a precipitate of light yellow needles of a very explosive compound occured, probably a sodium salt. Reacidifying the
precipitated putative sodium salt brings everything back into solution and the sodium salt can be reprecipitated by adding bicarbonate again. This
seems to exclude some obscure rearrangement taking place upon basifying.
Based on this, and after rereading patent US4246052, I'm reasonably sure the compound described in US4246052 and that obtained from the
copper/ascorbic reduction of styphnic are one and the same. Diazotisation in concentrated hydrochloric probably precipitates the chloride salt of the
diazoquinone due to the common ion effect. What remains strange is I have been unable to isolate the free diazoquinone itself, either it is extremely
soluble in water, or the pH range needs to be very strict. Maybe a different pka/b of the diazo group (relative to that of the two hydroxyl groups)
for 2-diazo 4,6-dinitro resorcinol compared to the 4-diazo derivative explains this, not sure.
Another observation was that the light yellow needles of the sodium salt, when acidified using acetic acid, did dissolve temporarily again,
precipitating small golden cubic crystals again that might be the free diazoquinone, not sure yet. It also detonates in reasonably small quantities,
so if it is the free diazoquinone, it behaves much different from 4-diazo 2,6-dinitro resorcinol, which only flashes upon ignition, like NC. It might
be made easily by adding the diazo chloride salt to acetic acid and slowly adding a sodium acetate solution. If this is all true, it might also be
interesting to see what the impact and friction sensitivity are compared to the salts, and 4-diazo 2,6-dinitroresorcinol.
4-diazo 2,6-dinitro resorcinol has been described by klapotke et al from a trinitro 4-aminophenol derivative. Describedly, it crystallizes from
reasonably concentrated nitric, as was also observed for the compound obtained by me from further nitration of 4-amino 2,6-dinitrophenol, which
therefore most likely also resulted in 4-diazo 4,6-dinitro resorcinol, as it was readily attacked by azides.
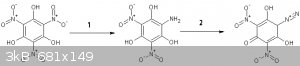
So it seems most likely that reduction of styphnic produces 2-amino-4,6-dinitro resorcinol and 4-diazo 2,6-dinitro resorcinol can only be obtained
from protected 4-aminophenol derivatives, similar to the routes to obtain 2-DDNP and 4-DDNP. Mystery solved I think! Traces of 4-diazo 2,6-dinitro resorcinol would show up after
reduction/diazotization, so why can ONLY the 2-nitrogroup of styphnic and picric be reduced, even by something as reasonably aspecific as SnCl2?
One other explanation could be the neat nitric or nitric/SA treatment results in some conversion back to stypnic (as has been show for picramic under
some conditions), resulting in some strange pi stacking adduct with styphnic, that did crystallize more easily from dilute nitric, but this is not
very likely IMO.
[Edited on 27-11-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Surprisingly, 2,4,6-tridiazocyclohexane-1,3,5-trione (Trisdiazo phloroglucinate) exists and can be made directly from phloroglucinol using diazo
transfer reagents (See 1st attachment), or trinitroso-phloroglucinol and hydrazine hydrate. (Attached article) Describedly, it detonates from heating
only at 220 deg C, so this could mean it is even more temperature stable as the DDNP's.
So, I don't see any direct reasons why a diazo dinitro phloroglucinate, or maybe a mononitro-bisdiazo phloroglucinate wouldn't be possible. Wondering
what could be the possible products for trinitrophloroglucinol, if the copper/ascorbic reduction is applied followed by HCl treatment. Guessing it
might be possible to produce 2-amino-4,6-dinitrophloroglucinol this way. Maybe
providing competing ligands for the Cu(I) (To keep the reduction products more solubilized) during the reduction and mild conditions, the mono-nitro
diamino phloroglucinol can be possibly made as well. Not sure how suscpible those amino groups would be to hydrolysis. Curious if these would be able
to produce diazo derivatives as well (2nd attachment). I'm guessing if they do, they would be completely undescribed diazo compounds!
The nitration of phloroglucinol kan be done under very mild conditions using KNO3/sulfuric acid IIRC, though where to get phloroglucinol, or
1,3,5-trihydroxybenzene?
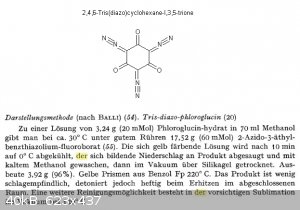
Attachment: Samsonov, V.A., Gatilov, Y.V. & Volodarskii, L.B. Russ Chem Bull (2012) 61 ; 1776.pdf (304kB)
This file has been downloaded 593 times
[Edited on 28-11-2018 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
An interesting possibility for a potential double salt may be a possible analogue for potassium DDNR with nickel styphnate, and it might form directly
if nickel carbonate could substitute for copper carbonate in the ascorbic acid reduction.
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Been doing some additional experiments involving styphnamic acid and it's diazotization, all is done on an extremely small scale (50-100 mg) due to
the hazards of these compounds, so not very precise. The solubility of the styphnamic acid in hydrochloric is very particular. Adding very
concentrated (30%) hydrochloric to the deep orange-red stypnamic, a light yellow precipitate is formed, probably of the chloride salt, that is nearly
insoluble in the concentrated HCl. When the mix was carefully diluted stepwise and weighted, the maximum solubility of styphnamic acid in HCl seems to
be at around 13-17% HCl. Further dilution results in a pretty sharp dissociation boundary, where the light yellow chloride salt transforms instantly
back to the orange-red free styphnamic acid. Since the solubility of styphnamic also depends on temperature and total volume of acid added, this would
be a lot of work to all sort out. It would also not be very meaningfull since the solubility of the diazoquinone in HCl seems to behave very different
as that of styphnamic itself.
Diazotization using 20% HCl at 0 deg C. produces scales or cubes of a very light yellow compound, probably the chloride salt as guessed earlier,
strange thing is that it seems to need 2 mole eqvts of nitrite in order the precipitate the light yellow chloride salt of the diazoquinone, but maybe
I'm just impatient...hmmm...weirdness... The putative chloride salt in itself
seems pretty explosive and large losses can occur due to the relatively high solubility of the diazoquinone in HCl. A better option seems
diazotization in 30-60% sulfuric. The sulfate salt seems less explosive as the chloride salt, presumably due to the bulkyness of the sulfate group and
recovery of the acid salt seems higher.
The potassium salt seems most interesting (if these are really salts and not the free diazoquinone), it is relatively insoluble in cold water and
shows higher brisancy compared to the sodium salt. It is easily made by neutralizing the chloride/sulfate salt using bicarbonate to almost neutral and
precipitation using saturated KCl. Upon recrystallization from hot water, the potassium salt precipitates as very fine light yellow needles clumping
togther. It behaves INCREDIBLY brisant, the tiniest specks blow holes in aluminium foil, like an azide or tetrazole derivative would. Also did a rough
explosion temperature measurement on my hotplate, surprisingly the explosion temperature seemed at least equal or even higher than that of the
2,6-dinitro 4-diazo resorcinol derivative, immidiate explosion temp. estimated around 250-260 deg C. Maybe due to higher purity.
Also tried the basic copper carbonate/ascorbic reduction for 3-aminopicric (3-amino 2,4,6-trinitrophenol) produced from the amination of picric acid
using hydroxylamine. The reduction resulted in an only 40% yield of some reduction product (after HCL treatment), noticably more brown in colour than
styphnamic. Melting point was similar to styphnamic though, as seemed the product upon diazotization. Wondering if this rather represents remaining
stypnic acid from the amination reaction, since it is done under very caustic conditions. Any thoughts on whether reduction of 3-amino
2,4,6-trinitrophenol is possible at all and if so...what would be the probable isomer(s) formed?
[Edited on 14-12-2018 by nitro-genes]
|
|
|
Tdep
National Hazard
Posts: 520
Registered: 31-1-2013
Location: Laser broken since Feb 2020 lol
Member Is Offline
Mood: PhD is done! It isn't good but it's over lol
|
|
| Quote: Originally posted by nitro-genes |
The potassium salt seems most interesting (if these are really salts and not the free diazoquinone), it is relatively insoluble in cold water
[Edited on 14-12-2018 by nitro-genes] |
Very interesting. Could you test if there is indeed potassium involved? Making up a sat solution in warm-ish water, then adding in some NH4/NaClO4,
should give you a clear positive test for potassium
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
That would indeed be a good idea, though I don't have any perchlorates available anymore. Experimenting with this putative diazoquinone on a very
small scale is really difficult, no idea actually what is the acid salt (if exist at all) the free diazoquinone and metal salts. If the diazoquione is
very acidic, the acid would just precipitate more due to less deprotonation, similar to picric being less soluble in dilute HCl. Been thinking that
the putative sulfate salt could be analyzed using calcium salts. At these scales though, adding 1 drop of something can mean the difference between
obtaining a precipitate or dissolving everything. For these kind of experiments you would also really need to wash things properly or perform at large
scale, both are rather difficult and/or dangerous.
Talking about perchlorates and dangerous....if the precipitate obtained after diazotization of styphnamic truly is an acid salt of the diazoquinone, I
wonder if a diazonium perchlorate would also exist and would precipitate, it would be like the ultimate primary from hell probably.  It is good that I don't have any perchloric acid, or would likely loose an eye or
some fingers.... It is good that I don't have any perchloric acid, or would likely loose an eye or
some fingers....
[Edited on 14-12-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
A test was done to determine if the compound isolated after diazotization of styphnamic is an acid salt or free diazoquinone:
Experimental:
0.5 g styphnamic was dissolved in 6 g of 50% sulfuric with slight warming, forming a very light yellow solution. Upon cooling to <0 deg C, there
was no precipitation of the sulfate salt (as can occur with concentrated HCl). Then, 0.17 g of sodium nitrite was added to 1.5 ml water and added
dropwise while in an icebath, some foaming was observed near the end of the reaction. Halfway the addition (about 15 minutes in), light yellow
crystals started forming. After stirring for another 60 minutes after the last addition, the light yellow crystals were filtered off, blotted dry as
best as possible using paper towels and dried for 8 hours at room temperature. Yield after drying was 0.49 grams. The dried precipitate is somewhat
flame sensitive, burning with an orange flame and sustains deflageration (although burnrate is very slow). Heated slowly to ignition point in 2-5 mg
amounts on a spatule, it never produces a detonation though.
The dried compound was cautiously transfered to a 20 ml beaker and destilled water was added dropwise at room temperature untill all dissolved. In
total, 0.82 grams of water (at room temperature) was needed. Then, 3 ml of a pH 7 buffered ~10% calcium acetate /acetic solution was added, total
volume adjusted to 10 ml, and kept at 4 deg. C. overnight. Only ~15 mg of calcium sulfate could be recovered after overnight at 4 deg C. Then, 1 ml
of the filtrate was transfered to a 5 ml beaker and a few drops of a near saturated KCl solution added while swirling. The solution immediately became
almost solid in apparance from abudant precipitation of light yellow needles of the potassium salt.
To the rest of the filtrate containing the very soluble calcium salt of 2-diazo 4,6-dinitroresorcinol was added a slight excess of ammonia. N2 gas was
evolved (quite slowly though) and long, light brown needles formed after 24 hours in the cold, presumably of 4,6-dinitropyrogallol or it's ammonium
salt. It burns only weakly energetic after drying.
The nitration of styphnamic was also attempted:
Experimental:
0.5 grams of styphnamic was added to 7.5 g 97% sulfuric acid. The styphnamic formed sticky lumps when contacting the sulfuric and was very hessitant
to dissolve, though eventually (and with a bit of persuasion) a light yellow solution was obtained. This was cooled to 0 deg C. in an ice bath and
slowly 0.6 g KNO3 was addded over the course of an hour or so. The light yellow solution became an incredibly dark red (like red wine). After stirring
for about 3 hours in total, the reaction was left on ice without stirring for another 12 hours. At this point the solution had become a light
yellow-orange. While still in the icebath, about 15 ml's of ice cold water was added dropwise. A lof of NOx was released. After another 8 hours or so
at -20C, yellow-orange and 3-5 mm long needles were present in the solution (judging by the colour, there might have been a lot more product left in
the filtrate, perhaps less water would need to be added to precipitate more). The needles were filtered off, blotted dry with paper towels and after
drying weighed about 100 mg. A few mg's on a spatule when heated slowly results in detonation. It also sustains deflageration from flame, flashing off
reasonably quickly, leaving a large black stain. All in all, it seems more energetic as the product obtained from diazotization of styphnamic using
nitrite/50% sulfuric above.
The rest of the product (~100 mg) was added to a 5 ml beaker, and distilled water again added until all dissolved. It needed 4.68 g of water to all
dissolve, so also it's solubility seems very different from that of the product obtained from direct diazotization. A bit of calcium carbonate was
added until the pH was neutral and a few drops of a saturated KCl solution again added. Upon cooling for several hours at 4 deg C., there was no
precipitation of a putative potassium salt. Solid KCl was added untill nearly saturated and cooled again. Small golden-orange cubic crystals of a
potasium salt appeared (attachment), which behaved very energetic. So, also the solubility and colour of the potassium salt seems different than the
product obtained from styphnamic diazotization. When touched by a glowing splint, it seems to make DDT in an even smaller amounts as the light yellow
potassium salt of 2-diazo 4,6-dinitroresorcinol itself.
Although it is quite possible that the nitration of styphnamic produced a small yield of 2-diazo 4,6-dinitrophenol from the nitrous produced, it is
tempting to speculate that a diazodinitrophloroglucinate resulted from the nitration by a putative 2-amino 4,5,6 trinitro resorcinol derivative!

[Edited on 21-12-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Ok, for everyone interested in repeating the experiments with 2-amino 4,6-dinitrophenol and diazotization products...Noticed I got a bitter taste in
my mouth from being in the same room as the drying diazo derivative. Some of the products like 4,6-dinitropyrogallol can sublime (temp unknown
though) and hardly anything about the potential toxicity of these compounds is known, so take adequate precautions when working with these compounds!
The bitter taste could be a coincidence, since I was recovering from the flu at that time, though it seemed better to at least mention this to others.
Wont be experimenting with these compounds myself anymore though.
[Edited on 27-12-2018 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
One more post then...
Initially, I had some trouble using hydrochloric acid to diazotize the styphnamic (2-amino 4,6-dinitroresorcinol) to the corresponding 2-diazo
derivative and determine whether this could possibly be an acid salt or the free diazoquinone. The free diazoquinone is very soluble in water and the
alternative use of 50% sulfuric makes it impossible to dry the end product. As posted before, the solubility of styphnamic acid behaves somehwat
peculiar in concentrated HCl (20-30%) and is prone to form a presumed acid chloride salt. The chloride salt of styphnamic is relatively insoluble at
lower temperatures and prevents the full dissolution of all styphnamic when cold concentrated HCl is used. During diazotization, this leads to
unreacted styphnamic in the end product.
Experimental:
0.25 grams of styphnamic acid was added to a 20 ml beaker. Then, 8 grams of 20% HCl was added and the solution heated slowly to about 90 deg. C.. All
the styphnamic went into solution, forming a light yellow solution. Upon cooling this to 0 deg. C. in an icebath, beautiful light yellow/golden
coloured leaflets started to form, presumably the chloride salt of styphnamic. Next, 0.1 g of sodium nitrite was added to a 5 ml beaker and 0.5 ml
water added to dissolve. While in the icebath and keeping temperature below 10 deg. C. the sodium nitrite solution was added over the course of about
20 minutes. Most of the chloride salt redissolved during diazotization. After stirring for another 2 hours in the icebath, the beaker was transfered
to a -20 deg. C. freezer and kept overnight. After filtering, the light yellow crystals were blotted dry as best as possible using filter paper and
dried at 50 deg. C. for 3 hours. No change in colour was observed during drying, total yield was 0.21 gram.
Unlike using sulfuric, the product from diazotization in HCl did seem to dry completely, and was very flame sensitive, behaving much like a
diazoquinone (videos attached). To test any possible presence of chlorides, 50 mg of the diazoquinone was transfered to a 5 ml beaker and a few drops
of water were added to dissolve. Adding a dilute silver nitrate solution did not produce any silver chloride (or other) precipitate. When a few drops
of a dilute HCl solution was added to the same solution afterwards, immediate clouding was observed. It seems the product is really 2-amino
4,6-dinitro resorcinol.
What remains strange is that I had heat tested the presumed diazoquinone from HCl diazotization before....I'm still 100% sure it detonated when a few
mg were heated on a spatule when last tested :S....very strange....The only differences were that (1) the styphnamic was freshly prepared, (2) the
precipitate after diazotization was filtered off after only a few hours and (3) a larger excess of sodium nitrite was used. Hmmm....there seems to be
quite some carbon produced as well upon deflageration, maybe it wasn't completely dry, or part of the diazoquinone decomposed during the prolonged
contact with HCl or upon drying at elevated temperature. The styphnamic was kept in an amber and airtight HDPE container at -20 in a freezer, so any
decomposition seems unlikely.
Attachment: 2-diazo 4,6-dinitroresorcinol 1.avi (1MB)
This file has been downloaded 807 times
Attachment: 2-diazo 4,6-dinitroresorcinol 2.avi (706kB)
This file has been downloaded 775 times
[Edited on 7-1-2019 by nitro-genes]
|
|
|
| Pages:
1
..
27
28
29
30
31
..
33 |
|








