| Pages:
1
..
23
24
25
26
27
..
33 |
PHILOU Zrealone
International Hazard

Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Quote: Originally posted by nitro-genes  | Thanks Philou, it seems the melting point is in range of what I found for the chlorinated benzoxazolone. Read something about it's use as muscle
relaxant, though I'm not inclined to eat anything I produce in my shed. 
Ok, here is something strange... This is based on 1 rushed experiment, it would
need further examination, but I'll add it here since I don't store these things on my computer. This is based on 1 rushed experiment, it would
need further examination, but I'll add it here since I don't store these things on my computer.
I was bored and had a lot of purified isopicramic acid left and decided to look at what exactly forms during nitration using 97% SA and a nitrate
salt. To a 20 ml beaker, 10 grams of 97% SA was added and 0.5 grams purified isopicramic acid added and stirred at 20 deg C until everything
dissolved, producing an almost red/black but transparent solution. Next, it was added to an icebath and at 0 deg C, 0.25 grams of KNO3 (~1 mol eqvt)
was added. The solution was allowed to stir and no gas evolution was seen. Gradually, the solution took on a much lighter red transparent colour,
indicating something was happening. A small sample withdrawn at this stage dissolved in icecold water completely, not a single bubble of gas was
produced. Since this could be exlained by the H2SO4 adduct with isopicramic itself, I decided to add another mole equivalent of KNO3 and very soon,
gas formation became evident. It was allowed to stir overnight in the icebath, going from 0-10 degrees, with steady evolution of gas. When water was
added in the morning, copious amounts of NOx were liberated and a transpararent yellow/orange solution was left. No precipitate occured when kept at 4
deg C overnight. About 3 ml's of ethyl acetate were added and briefly stirred, upon which almost all of the colour transfered to the organic phase.
This was siphoned off and allowed to evaporate. A small amount of a yellow/orange crystalline precipitate formed, that burned very characteristic for
a diazonium compound, very vigorous (more than p-DDNP) and with yellow flash. It dissolved very easily in water again, but adding a saturated KNO3
solution and chilling produced no precipitate. It further seems to attack metals like crazy, although this could also be due to some extracted acid by
the ethylacetate.
So, what formed here? Since it is water soluble, even after extraction, it is
not p-DDNP iself, since the diazonium sulfate salt dissociates very quickly upon dilution. It still contains a diazonium group, but since it does not
produce a precipitate with KNO3, it likely also isn;t DDNR, since the K-salt is reported to be very insoluble (DDNR istelf as well). One of the
options is that a 1,2 quinone 3,6 dinitro 4-diazo is formed due to hydrolysis of the 2-nitro of isopicramic acid, though I'm not sure this would
explain the reactivity towards metals. Another option is the 2,3,6 trinitro 4-diazo phenol descirbed before, though this would be strange considering
the described deactivation of the amine group in 97% SA. Other IMO, less likely options would be some sulfonic acid replacement of the nitro, or more
likely maybe some azoxy compound from coupling reactions. Any guesses, anyone?
[Edited on 17-12-2016 by nitro-genes] |
Answer is hard to give   as usual
as usual 
You have initially H2N-C6H2(NO2)2-OH probably as a sulfate.
Adding KNO3 results into liberation of some HNO3.
-->Part of the HNO3 may oxydise the para-amino-phenol into a para-quinon (mono-imine)...
-->Another part may nitrate further the dinitro compound into a trinitro one...or doubtfully to a tetranitro one
I don't think so, but, if it does, then you will have two NO2 on neightbourgs C (ortho vs each other and ortho vs the NH2 for one and vs the OH for
the other)...and in such conformation; one of the two neightbourgs NO2 may be expandable/lost by hydrolysis afterwards and lead to even more oxydable
molecules (dihydroxyaminobenzene and by further hydrolysis to trihydroxybenzenes (or trihydroxyaminobenzene to tetrahydroxybenzene).
Oxydation of the substrate by HNO3 is concomittant with reduction of HNO3 into NxOy fumes in what nitrosonium is present as NO(+)...a powerful
nitrosating agent.
I have seen into a book about diazocompounds a process to make diazoniums starting from an aniline and plain HNO3 only; thus without any
nitrite...about half of the HNO3 serves to make the oxydation and NO(+); the other half is used to make the salt diazonium nitrate.
Usually para- and ortho-aminophenol when diazotized makes an unsoluble internal diazonium-oxyde (diazo-phenate); but here maybe the H2SO4 keeps it
solubilized? And then it reacts further with unoxydized p-amino-phenol molecule to make a triazene...or a quinone based Schiff's base.
The reaction scheme here-below is simplified since all ortho- and para-quinon (and imines) derived from transcient trinitro and tetranitro derivatives
by hydrolysis/oxydation may also react
--> with the NO(+) to make diazoniums
--> or with the free p-amino-phenol molecule to make triazenes or quinon based Schiff's bases.
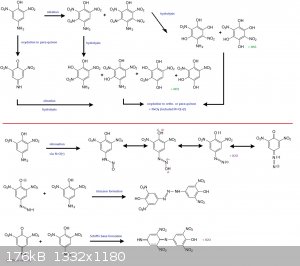
[Edited on 18-12-2016 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Best wishes for 2017 everyone! Thanks for that scheme Philou, there is probably a lot happening as indeed is usually the case.
So here are my further incoherent ramblings regarding the subject :
When the product after KN/SA nitration was extracted with ethylacetate, redissolved in a minimum of 80 deg C water, pH adjusted to 6 and KNO3 added, a
light yellow-orange coloured compound precipitated, contaminated by a brownish-orange compound, which is most likely pDDNP. The light yellow-orange
coloured material is incredibly explosive though, the tiniest specks detonate with considerable strength, much resembling a heavy metal azide. It
concurs with desricptions of the potassium salt of DDNR, although a potassium salt of a 1,2 quinone diazide is also possible. Total yield was only
about 100 mg or so, so for curiosity purpose only. Most of the ethylacetate extracted stuff is lost when redissolved, so I guess that the 1,2 and 1,4
quinones and accompanying diazo derivatives therefrom are likely far more water soluble than pDDNP and DDNR. The compound could also be a potassium
salt of the triazene as indicated in your scheme, though I don't think this is likely under these extremely acidic conditions. Likely, the nitramine
is formed first after which several things could happen. Also don't think direct NOx mediated oxidation to the quinone imine is likely under these
acidic conditions. When I dissolved purified isopicramic acid in boiling (!) 30% sulfuric acid once, and added potassium nitrite in small portions,
almost exclusively pure, crystalline pDDNP was produced as large coarse crystals. The reaction was much slower in this case, likely due to the fact
that the anilinium ion is unreactive towards NOx, only the small amount of dissociated and free acid reacting. Then again, these temperatures would
also lead to fast decomposition of the HNO2 and N2O3 produced, which would be the main diazotizing species in this case.
Still puzzled why heating 2,3,6-trinitro 4-aminophenol with 65% nitric produces DDNR selectively and in such good yields. Maybe this is a tradeoff
which allows hydrolysis of the 3 nitro, enough water to allow diazotization by the NOx produced (or nitramine formation) while relatively disfavouring
further nitration of the ring? It is strange though, both DDNR and 2,3,6-trinitro 4-aminophenol are reported to be reasonably stable in cold
concentrated sulfuric acid (no hydrolysis there), so yields must be bad for other reasons, maybe using sulfuric acid changes the way the nitramine
rearranges or reacts, though I would have guessed ring nitration (if possible) would be favored this way. I was unable to find how a diazonium salt or
diazophenol behaves in concentrated sulfuric acid but maybe some side reactions occur here as well. Also curious whether using 100% SA or even oleum
as opposed to 97% SA would have made any difference, or 2 mole equivalents of 100% HNO3 in an inert solvent or something. Or similar nitration using 1
mole eqvt of nitrate salt.
Below is 1 mg of the putative potassium salt of DDNR on aluminium foil (Dornier style). Pretty hot stuff, destroyed the rest...
Attachment: 1 mg Potassium salt of DiazoDiNitroResorcinol vid - Copy.avi (2.8MB)
This file has been downloaded 1046 times 
[Edited on 3-1-2017 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes | Best wishes for 2017 everyone! Thanks for that scheme Philou, there is probably a lot happening as indeed is usually the case.
So here are my further incoherent ramblings regarding the subject :
When the product after KN/SA nitration was extracted with ethylacetate, redissolved in a minimum of 80 deg C water, pH adjusted to 6 and KNO3 added, a
light yellow-orange coloured compound precipitated, contaminated by a brownish-orange compound, which is most likely pDDNP. The light yellow-orange
coloured material is incredibly explosive though, the tiniest specks detonate with considerable strength, much resembling a heavy metal azide. It
concurs with desricptions of the potassium salt of DDNR, although a potassium salt of a 1,2 quinone diazide is also possible. Total yield was only
about 100 mg or so, so for curiosity purpose only. Most of the ethylacetate extracted stuff is lost when redissolved, so I guess that the 1,2 and 1,4
quinones and accompanying diazo derivatives therefrom are likely far more water soluble than pDDNP and DDNR. The compound could also be a potassium
salt of the triazene as indicated in your scheme, though I don't think this is likely under these extremely acidic conditions. Likely, the nitramine
is formed first after which several things could happen. Also don't think direct NOx mediated oxidation to the quinone imine is likely under these
acidic conditions. When I dissolved purified isopicramic acid in boiling (!) 30% sulfuric acid once, and added potassium nitrite in small portions,
almost exclusively pure, crystalline pDDNP was produced as large coarse crystals. The reaction was much slower in this case, likely due to the fact
that the anilinium ion is unreactive towards NOx, only the small amount of dissociated and free acid reacting. Then again, these temperatures would
also lead to fast decomposition of the HNO2 and N2O3 produced, which would be the main diazotizing species in this case.
Still puzzled why heating 2,3,6-trinitro 4-aminophenol with 65% nitric produces DDNR selectively and in such good yields. Maybe this is a tradeoff
which allows hydrolysis of the 3 nitro, enough water to allow diazotization by the NOx produced (or nitramine formation) while relatively disfavouring
further nitration of the ring? It is strange though, both DDNR and 2,3,6-trinitro 4-aminophenol are reported to be reasonably stable in cold
concentrated sulfuric acid (no hydrolysis there), so yields must be bad for other reasons, maybe using sulfuric acid changes the way the nitramine
rearranges or reacts, though I would have guessed ring nitration (if possible) would be favored this way. I was unable to find how a diazonium salt or
diazophenol behaves in concentrated sulfuric acid but maybe some side reactions occur here as well. Also curious whether using 100% SA or even oleum
as opposed to 97% SA would have made any difference, or 2 mole equivalents of 100% HNO3 in an inert solvent or something. Or similar nitration using 1
mole eqvt of nitrate salt.
Below is 1 mg of the putative potassium salt of DDNR on aluminium foil (Dornier style). Pretty hot stuff, destroyed the rest...
[Edited on 3-1-2017 by nitro-genes] |
@ Nitro-genes ... Beste wensen! Vroolijke nieuwe jaar!
@ The rest of the world ... May all your wishes come true! Happy new year!
Nice video-tje!
Still puzzled why heating 2,3,6-trinitro 4-aminophenol with 65% nitric produces DDNR selectively and in such good yields. Maybe this is a tradeoff
which allows hydrolysis of the 3 nitro, enough water to allow diazotization by the NOx produced (or nitramine formation) while relatively disfavouring
further nitration of the ring?
Probably by the reason I exposed about the making of diazonium nitrate from anilin and HNO3...but with one more subtility
The 2,3,6-trinitro-4-amino-phenol is first oxydised partially into 2,3,6-trinitro-4-imino-benzoquinone by HNO3 and generating subsequently HNO2.
HNO2 turns the rest of the 2,3,6-trinitro-4-amino-phenol into 2,3,6-trinitro-4-diazo-phenate.
The later 2,3,6-trinitro-4-diazo-phenate display synergetic effect of the diazonium (meta director) in position 4 and of the two nitro (meta director)
from position 2 and 6...all working against the NO2 in position 3 (a bit the same as having 2,3,4,6-tetranitrophenol)...which rearranges into nitrite
what is readilly hydrolyzed by the water arround and the boiling providing a -OH group and fresh N=O(+) as (HONO).
Ar-NO2 <--==> Ar-O-N=O (forced by ortho and para EWG groups)
Ar-O-N=O + H-OH <--==>Ar-O-H + HO-N=O
Thus the subtility here is that only a tiny amount of nitrite is needed to persue the full diazotation because it is regenerated by the
hydrolysis step...and acts a bit like a catalyst.
For the rest you get more questions/interogations than answers...vive la science
Let's hope you "destroyed the rest" with the due timing arround midnight amongst the explosions of the new year fireworks .
[Edited on 3-1-2017 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks Philou, that would indeed be as described by Meldola and reverdin and in later publication by Atkins and Wilson. That procedure you mention
about making diazophenols from aniline would be interesting to read btw!
Out of curiosity I set up 2 new experiments involving the nitration of isopicramic acid both using 97% SA and KNO3:
To 2 separate 50 ml beakers were added 15 grams of 97% sulfuric acid and cooled to -20. To the first beaker was added 0.55 grams of KNO3 (1.1 mole
eqvt) and to the other 1.1 grams of KNO3 (2.2 mole eqvt). Both were chilled to -20 deg C after which 1 gram of purified isopicramic acid was added
with good stirring. No ice cooling was used, the reactions were allowed to stir at ambient temperature (~10 deg C.) for 12 hours in a water bath,
after which about 15 m's of water were added. When water was added to the beaker containing 1.1 mole eqvt of KNO3, some NOx escaped and after a short
while, crystalline pDDNP started to crystallize, with bright yellow supernatant.
When water was added to the beaker containing 2.2 mole eqvt of KNO3, lots of foaming occured, leaving a dark orange liquid, with no precipitate. The
liquid was extracted using about 10 ml's of ethylacetate and added to a 100 ml beaker. The beaker containing the ethyl acetate extract was heated to
50 deg C. and allowed to evaporate nearly completely. During evaporation, a crystalline, bright yellow compound started to precipitate. Only when
nearly all of the ethyl acetate was evaporated, an additional dark orange compound also seemed to precipitate. When all ethyl acetate was evaporated,
10 ml of cold water was added and heated with stirring to 80 deg C. When about 40-50 deg C was reached, the solution started to fizzle, NOx was
released and a yellow-orange precipitate occurred, with dark orange supernatant. The contents were filtered and heated to 80 deg C. in 10 ml of fresh
dH2O, 1 gram KNO3 was added and allowed to cool to 4 deg C. About 300 mg of a yellow-orange crystalline precipitate occured, behaving much like a
heavy metal azide. After second thought, based on the colour and water solubility, I would say the compound is most likely the potassium salt of
2-oxide 3,6 dinitro 4-diazophenol.
Very curious what exactly the bright yellow compound after ethyl acetate evaporation is. It does seem to react with water releasing NOx and is highly
soluble in ethyl acetate. The beaker containing 1.1 mole eqvt of KNO3 produced mostly pDDNP, this would be consistent with simple dissociation of a
diazonium sulfate or hydrolysis of a nitramine , and not a trinitro compound though. This makes an interesting question why 2.2 mole eqvts of KNO3
produces a different product though. Either p-DDNP is nitrated further, or the primary nitramine is nitrated further, perhaps by NyOx present,
perhpaps more like a radical nitration. Or the compound is a trinitro nitramine, which hydrolyses expelling one nitro group and leaving a diazo as has
been shown before (nitration of 3-nitro aniline to tetranitronitranline --> O-DDNP), this would make the compound more likely to be DDNR though. If
the compound would be a trinitro nitramine, it would be interesting to repeat this experiment using 100% sulfuric acid maybe or even oleum to prevent
premature diazo formation and oxidation side reactions, very similar to why only oleum can be used to produce tetranitro aniline. If it is more like a
radical nitration, less product should be obtained, or even only pDDNP maybe?
[Edited on 5-1-2017 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Yes stange that you get two different reactions by working with a double KNO3 dose.
Maybe you finally got p-DDNR via 2,3,6-trinitro-4-aminophenol or via 2,3,6-trinitro-4-diazophenate?
I highly doubt any polynitro-aromatic nitramine could be present here...
Because:
Two or more nitro groups increases a lot the acidity of the nitrogen protons onto an aromatic ring...and it is known that above a certain pKa the
nitrogen (-NH-) can't hold a NO2 long ... especially if water is present ...
See weak hydrolytic stability towards water of dinitrourea or of dinitramide
EWMP-NH2 + NO2(+) <==--> EWMP-NH-NO2 + H(+) (EWMP = Electron Withdrawing Molecular Part)
Would be nice to test p-DDNP against water stirring to 80°C and see if arround 40-50°C there is also fizzing and NOx release...that way you exclude
the fizzing to come from thermo-hydrolytic-decomposition of a diazo-oxyde (diazophenate).
The introduction of a 4th nitro-group into trinitroaniline only requires oleum because the aromatic ring becomes very electron deficient and that it
requires more forcing NO2(+) conditions to suceed...the oleum doesn't play in that process an "anti-oxydizing" role.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Very exiting, just had to make a post!
Assuming that the intermediate upon nitration of isopicramic is indeed a trinitrodiazoxy, and some intermediates may not be stable for prolonged time
in sulfuric acid I changed the procedure somewhat:
10 grams of 97% sulfuric acid was cooled and 1.05 grams of KNO3 was added and stirred to dissolve. This was cooled to -20 and 1 gram of purified
isopicramic acid was added at once. This was allowed to stir in an icebath for 4 hours, yielding a clear orange red solution, only slight gas
formation occured. After 4 hours, about 0.5 ml of water was added while still in the icebath, NOx escaped and this mixture was stirred for another 30
minutes on ice to allow diazotization, untill a sample withdrawn produced a light yellow precipitate. Slowly, not letting temperature rise, more water
was added and 3 ml ethylacetate to extract the dark orange stuff. To my delight, the bottom of the 20 ml beaker is now covered with a good layer of
light yellow, almost transparent glittering crystals. Hopefully the ethylacetate didn't precipitate KHSO4 or something, or this is some differently
coloured pDDNP crystal variant or pDDNP sulfate.
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
Calling Dr. Klapotke ...structural theory / resonance hybrid / NMR anomaly ahead....
time to get your freak on
Maybe we should drill down a bit further and see what we find huh

[Edited on 1/7/2017 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
It'aint rocket science, but with the extended laws on prohibition of HE related chemicals over here (AN, conc. nitric, (per)chlorates, nitromethane
etc), dumping some isopicramic acid in rootcleaner with KNO3 is at least still somewhat legal over here, besides I wanted to get rid of the
isopicramic acid left as well. These are just not the times anymore for amateur chemistry, especially HE related... Maybe sometime again...thank you
guys for all the help!
Last overview of the experiments done last couple of weeks:
0.5g pDDNP --> suspended/dissolved in 5 grams 97% SA, dark red black colour --> 4h 10 deg C. (no gas formation, no change in colour) -->
Adding ice --> no gas formation, immediate dissociation (or precipiation) --> crystaline pDDNP
0.5g Isopicramic --> dissolved in 5 g 97% SA, transparent dark red colour --> 4h 10 deg C. (very little gas formation, no change in colour)
--> Adding ice --> very little gas formation, no smell of NOx, yields transparent red solution with some minor dark red flocculent material.
Adding solution of nitrite yields crystaline pDDNP, with minor amounts of dark brown material
1 g Isopicramic --> dissolved in 10 g 97% SA + 2 mole eqvt of KNO3 --> 4h 10 deg C. (some gas formation, smell of NOx, colour changes from dark
transparent red, to very dark red, to light orange red) --> adding water --> lots of gas formation, smell of NOx, upon cooling precipiation of
~0.3 grams of light yellow needle shaped crystaline material + small amount of dark orange/red ethyl acetate soluble stuff. The light yellow material
is almost water insoluble and acidic. Adding bicarbonate and heating until near neutral dissolves most of the compound with dark yellow colour,
seemingly precipitating some pDDNP similar material, over neutralizing leads to dark brown solution and no precipitation.
1g pDDNP --> suspended in 97% SA + 1 mole eqvt of KNO3 --> 4h 10 deg C. (some gas formation, colour changes from dark black-red to transparent
red. --> adding water --> Some gas formation, smell of little NOx -> precipiation of large amount of unchanged pDDNP, dark yellow orange
supernatant. Decanting and heating solution causes more NOx formation and precipitation of small amount of light yellow material. Light yellow
compound is acidic in water and burns characteristic for diazo compound, leaving significantly more black residue though.
0.5g p-DDNP --> dissolved/suspended in 10g 97% SA + 2 mole eqvt of KNO3 --> heating (40-50 deg C) untill all dissolves --> very fast change
to orange-yellow colour, very little gas formation, very faint smell of NOx --> 1h 40 deg C. no further colour change--> adding water -->
very slight gas formation --> upon cooling, large amount of the light yellow crystalline precipitate.
Baffled regarding the identity of the light yellow compound. Difficult to tell, but during the reaction of pDDNP with KNO3, the reaction happens too
fast and there seems too little N2 escaping to explain the formation of a triazene. The burn characteristics and extra amount of residue produced on
burning would however be consistent with the triazene from Philou's scheme. Other options are a very differently coloured crystal variant of pDDNP
(that can be reversed by boiling in water), or some really weird double salt /adduct/complex of p-DDNP with something. Either way, it seems that the
small amount of the extremely explosive diazo compound obtained earlier occurs through direct ring nitration of isopicramic, which yield is probably
only slightly increased when performing the reaction in 100% SA.
Attachment: Light yellow mystery compound - Copy.avi (1.3MB)
This file has been downloaded 1010 times
[Edited on 8-1-2017 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
I forgot one point that puzzle me about diazoniums...and that I should try one day.
Picric acid, trinitroanilin and related compounds (2,4-dinitrophenol pKa= 4,09) are acidic...so there is also a chance to make a diazonium picrate.
If one puts for example water solutions of picric acid, NaNO2 and dinitroanilin into a single recipient...one should see...
sodium picrate form but also a putative dinitrodiazonium picrate...
NaNO2 + HO-C6H2(NO2)3 --> NaO-C6H2(NO2)3 + HNO2
HNO2 + H2N-C6H3(NO2)2 --> HO-N=N-C6H3(NO2)2
(O2N)3C6H2-OH + HO-N=N-C6H3(NO2)2 --> (O2N)3C6H2-O-N=N-C6H3(NO2)2 + H2O
There is also a chance some extra picric acid stack onto it via complexation.
I haven't taken trinitroanilin into my example because it is following pKa tables more acidic than TNP.
But thinking about it, it would be worth to allow trinitroanilin to react with 1/2 equivalent of NaNO2...
H2N-C6H2(NO2)3 + NaNO2 --> NaHN-C6H2(NO2)3 (*) + HNO2 --> Na-O-N=N-C6H2(NO2)3 (**) + H2O
(O2N)3C6H2-NH2 + Na-O-N=N-C6H2(NO2)3 --> (O2N)3C6H2-NNa-N=N-C6H2(NO2)3 (***) + H2O
I wonder about the primary abilities and explosive properties of trinitroanilinates (*), trinitrodiazotates (**) or bis-trinitrophenyl-triazenates
(***)...
On another hand 2,4,6-trinitroanilin when diazotized is apparently (from what I have understood in one of my readings) subject to hydrolysis of one of
its ortho NO2 via nitro-nitrite rearrangement (thus substituted by a OH) and leads to another (iso-)o-DDNP variant (2-diazo-3,5-dinitro-phenol) to
compare with conventional o-DDNP (2-diazo-4,6-dinitro-phenol)...so maybe no trinitroanilinates (*), trinitrodiazotates (**) or
bis-trinitrophenyl-triazenates (***)...and only iso-ortho-DDNP.
So the DDNP tread is far from dead...stil a lot of research to do for all SM members
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
A diazonium picrate/2,6 dinitrophenate would certainly fit the bill, being titratable with weak base with recovering of some pDDNP. I must say, I've never seen any precipitation of picric using long nitration times of
isopicramic though, puzzling...Would be easy enough to weigh the yellow product and pDDNP recovered and see what is present in the supernatant after
dissociation. Picric acid, 2,6 dinitrophenol or perhaps KHSO4. Usually, more forcing conditions are needed to remove the diazo group IIRC, especially
expect so for pDDNP, but maybe I'm wrong there and part of the diazonium is replaced by a nitro or eliminated. Such a diazonium salt would in any case
be remarkable considering the presence of SA, which is far more acidic than even picric, so maybe it would be largely solubility driven or more like a
complex?
Where did you read about the hydrolysis of the 2-nitro of picramide btw? Nitration of picramide using NA/SA yields nothing but starting material IIRC,
and only under special conditions (Conc SA or PPA, or something) a diazonium salt can be obtained. Boiling in alcohol of the latter yields mostly
trinitrobenzene from what I've read, reaction with water picric.
[Edited on 8-1-2017 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes |
Where did you read about the hydrolysis of the 2-nitro of picramide btw? Nitration of picramide using NA/SA yields nothing but starting material IIRC,
and only under special conditions (Conc SA or PPA, or something) a diazonium salt can be obtained. Boiling in alcohol of the latter yields mostly
trinitrobenzene from what I've read, reaction with water picric.
|
Into the following book:
Traité de chimie organique-Tome XV - Masson & Cie Editors
by G.Dupont, R.Locquin
Under the direction of V.Grinard and as secretary Paul Baud
Tittle:
Diazoïques et azoïques - triazènes, tetrazènes et pentazdiènes - azoxydérivés - hydrazines - hydroxylamines - oximes - amidoximes - azides
815 pages of pure delightfull informations all in french
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Nice, sounds like an interesting book to read (a translation that is)! Curious what procedure they used going from TNA, could you give a short
outline?
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
I did a quick search into my book and found the main reference to what I exposed, although it is not strictly speaking 2,4,6-trinitroanilin
(picramide) but the reaction must be the same with it because it involves 2,4-dinitroanilin and 2-nitroanilins.Here follow some pictures of pages 129
to 138.
The information is not straight because there are a lot of condensed information into various places of the book and even if I'm french speaking, I
have to read it twice or more to fully understand the potential of those condensed and generalised informations (and avoid misunderstandings)...too
much scientific condensation may lead to this.
I will try to translate it.









PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks for that, only scanned through the reaction schemes, translation would be appreciated! Interesting reactions, for the 1-amine-2-chloro-3 amine
(bottom 6th document from top), and the topic of halogen migration, some very complex chemistry there. I think the position of the nitrogroups ortho/para repsective of the amino doesn't favor any hydrolysis/substutions
and H-bonding of those aminohydrogens to the nitrogroups adds a lot of stability to TNA (and steric hindrence for amino), it's diazonium salt being
only obtained with much difficulty and the diazogroup itself most liable to decomposition, being sandwiched between two very strong EWG groups. Would
be interesting to hear your ideas though.
I was wrong about tetranitroaniline to o-DDNP conversion, it was 2,3,5 trinitroaniline shown to behave like that because of liable 2 nitro and
(presumably, maybe?) absence of additional 6 nitro.
[Edited on 9-1-2017 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
| Quote: Originally posted by nitro-genes | Thanks for that, only scanned through the reaction schemes, translation would be appreciated! Interesting reactions, for the 1-amine-2-chloro-3 amine
(bottom 6th document from top), and the topic of halogen migration, some very complex chemistry there. I think the position of the nitrogroups ortho/para repsective of the amino doesn't favor any hydrolysis/substutions
and H-bonding of those aminohydrogens to the nitrogroups adds a lot of stability to TNA (and steric hindrence for amino), it's diazonium salt being
only obtained with much difficulty and the diazogroup itself most liable to decomposition, being sandwiched between two very strong EWG groups. Would
be interesting to hear your ideas though.
[Edited on 9-1-2017 by nitro-genes] |
The case of TNA is not clear...and confusing because:
1°) it indeed needs more forcing conditions to diazotize thus one may think it is unstable and poor yielding.
2°) the resulting diazonium is very reactive (reacts faster and easier than other diazoniums) thus one may think it is less stable.
3°) it is stable at much higher temperatures (>40°C) than all other diazoniums (no need for an ice bath for example) thus one may think it is
more stable.
4°) the solid salt is more sensitive and explosive than any other thus one may think it is less stable...but maybe only the result of 3 nitro's.
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Point 1 maybe due to the need for strong concentrated acids in order to dissolve all of the TNA as the anilinium ion maybe and keep all of the
diazonium formed as an acid salt. Or maybe the diazotizing species is not nitrosylsulfuric, only from 80% sulfuric N2O3 is formed in significant
amounts IIRC, and even then this may have a hard time reaching the amino group due to steric hindrance. On the other hand I've also read that this may
require long reaction times for diazotization, since the aniliunium ion is unreactive towards diazotization. Don't know which is more correct or
maybe both are correct. In any case, this may indeed be more related to the solubility of TNA, diazonium salt, stability and reaction kinietcs.
Point 3 is interesting, maybe the increased reactivity is explained also here by more ready dissociation of the diazonium salt during further
reactions, which may not be the case for the solid salt at higher temperatures.
[EDIT] That reaction of the 2,4 dinitrodiazonium with NaOH (first page) is really interesting! Curious what conditions were used, as I would have
expected the alkalinity required to hydrolyse one of the o-nitro's would also immediately destroy the formed diazophenol as well. Is that diazophenol
much more stable? Did they slowly titrate the diazonium with base. Would sodium bicarbonate also work? The same reaction may be interesting for TNA
and tetra nitroaniline as well, though my gut feeling would be no o-nitro hydrolysis there.
Another random thought would be that the obsered light yellow colour for the K-DDNR salt in one of the patents earlier posted might relate to the same
light yellow precipitate containing pDDNP. Since they use KNO3-SA mediated diazotization, this would be consistent with an KHSO4 diazonium salt, which
upon recrystallization would yield the brown dissociated K-DDNR again. Assuming no decomposition during recrystalization, this would make sense.
[Edited on 10-1-2017 by nitro-genes]
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Here comes a fast translation of the previous 10 pages I have posted as images...took me more time  because text treatment structure is lost when copy-pasting from word to the forum text protocol. because text treatment structure is lost when copy-pasting from word to the forum text protocol.
In red personnal comments or additions to make it more clear.
In green the page cuts.
In blue the provided examples in text instead of structural formulas.
----------------------------------------------------------------------------------
p 129
Elimination, substitution or migration of groups into diazonium compounds
Elimination or substitution
The transormation of the amine function into a diazo one impacts strongly the stability of groups present onto the aromatic ring.
We will see that electro-negative groups provide stability to diazonium salts of strong acids; but it must be noticed that, if those diazonium salts
are treated by an alkaline agent or by an alkaline metal salt of a weak acid, then the electro-negative group becomes particularly labile; it is slit
off and replaced by a -OH group, what with the diazonium function, will cyclize into a diazo-oxide.
Those substitutions can occure during the diazotation process itself when a weak acid is present, and in that case, one gets not the expected
diazonium salt but the same diazo-oxide as one would get from performing diazotation onto an amino-phenol.
example:
1-diazonium 2,4-dinitro-benzen salt + NaOH provides the same as 2-amino-4-nitro-phenol + HNO2 = 2-diazo-4-nitro-phenoxide
Those group replacement phenomenons have been wel studied by Meldora and his students into the case of polynitroanisidins; we will spend a little time
on those works to allow for a better understanding of the large number of cases that we will further examine.
Study of the polynitro-anisidins.
If we take a look at the different dinitro-anisidins, we notice that when diazotized in the presence of sulfuric acid, they yield the normal diazonium
sulfate; but that when diazotized in the presence of chlorhydric acid, one of the NO2 groups in ortho vs the NH2 one is replaced by Cl; and finally
when a weak acid is present, like acetic
----------------------------------------------------------------------------------
p 130-131
Table: Diazotation of some polynitro-anisidins in presence of a weak acid
para-anisidins and one p-amino-phenol (missing data 4-amino-3,5-dinitro-anisole)
Line 1: 4-amino-2,6-dinitro-anisole eliminates the methyl to yield para-DDNP (2,6-dinitro-4-diazo-phenoxide)
Line 2: 4-amino-2,5-dinitro-anisole eliminates the methyl to yield another para-DDNP (2,5-dinitro-4-diazo-phenoxide)
Line 5: 4-amino-2,3-dinitro-anisole eliminates the 3-NO2 to yield 6-diazo-2-nitro-3-methoxy-phenoxide
Line 7: 4-amino-2,3-dinitro-phenol makes another para-DDNP (2,3-dinitro-4-diazo-phenoxide)
Conclusion from line 5-7 -OH is leading over -OCH3
ortho-anisidins (missing data 5,6-dinitro; 3,5-dinitro; 4,6-dinitro and 3,6-dinitro)
Line 3: 2-amino-4,5-dinitro-anisole eliminates the 5-NO2 to yield 4-diazo-2-nitro-5-methoxy-phenoxide
Line 4: 2-amino-3,4-dinitro-anisole eliminates the 5-NO2 to yield 4-diazo-2-nitro-5-methoxy-phenoxide
meta-anisidin (missing data 2,4-dinitro; 2,6-dinitro; 2,5-dinitro; 4,5-dinitro and 5,6-dinitro)
Line 6: 3-amino-4,6-dinitro-anisole forms a normal diazonium…no elimination stange
because 2,4-DNA does
----------------------------------------------------------------------------------
p 132
acid, then a methoxy group or a nitro group is replaced by -OH and subsequent formation of a diazo-oxyde (1).
Meldora has established that to be labile, the group must be placed in ortho or para position of the diazotizable amine function but also that it must
be kind of activated by the presence of a NO2 group in ortho position next to it (2). So a methoxy group -OCH3 in meta position vs the NH2
is not exchangeable even if next to a NO2 group.
In the case where two groups -OCH3 and -NO2 fulfill simultaneously those conditions; then it is the later that is favored for the splitting away.
Those reactions does not occure when the ring is holding a free -OH group in ortho or para of the function -NH2 (3); in that case indeed,
the stable diazo-oxide form is generated without any elimination.
Those rules are demonstrated by a few examples gathered into table at page 130 (and 131).
Noticing that when the -NO2 group is split away it turns into nitrous acid form, Meldora was able to persue diazotation of those nitro-anisidins with
about 1/4th of the theorical quantity of sodium nitrite normaly required (4); indeed this quantity starts the diazotation and the acid HNO2
generated by elimination of the -NO2 group, is enough to allow it to go on.
Elimination of the methoxy group.
Other cases than those of the polynitro-anisidins are known, in particular those of the nitro-anisic sulfonic acids.
Here also the -OCH3 group must be placed in ortho or para of the amine function and be activated by an ortho -NO2 group. It is the case to get to the
following diazo-oxides (5):
2-amino-6-nitro-4-sulfonic-anisole turns into 2-diazo-6-nitro-4-sulfo-phenoxide
4-amino-6-nitro-2-sulfonic-anisole turns into 4-diazo-6-nitro-2-sulfo-phenoxide
Elimination of the nitro group.
In a more general way than for the polynitro-anisidins, amines that are nitrated ortho vs the NH2 give rise to elimination of the -NO2 group during
diazotation if the para position is occupied by an electro-negative group.
Into the benzen family, 1-amino-2,4-dinitrobenzen (6), 1-amino
----------------------------------------------------------------------------------
p 133
-2-nitro-4-sulfo-benzen, 1-amino-2-nitro-4-arseno-benzen (7) , 1-amino-2-nitro-4-chloro-benzen (8),
1-amino-2-nitro-4-chloro-5sulfo-benzen (9), all lose their -NO2 group placed in ortho vs the -NH2 and lead to the formation of the
corresponding diazo-oxide by diazotation in acidic media. (weak or strong acid?)
1-amino-2-nitro-4-chloro-benzen turns into 2-diazo-5-chloro-phenoxide
Same kind of reaction process is observed into the naphtalen family. So 2-amino-1,6-dinitro-naphtalen diazotized in presence of concentrated sulfuric
acid and then dilluted by water immediately delivers the diazo-oxide (10):
2-amino-1,6-dinitro-naphtalen tuns into 2-diazo-6-nitro-naphtyl-1-oxide
The same happens to 2-amino-1,8-dinitro-naphtalen and to 1-amino-2,4-dinitro-naphtalen (11).
Halogen elimination.
Meldora observed that not only -NO2 and -OCH3 groups but also halogen ring substituants become labile by treatment of diazoniums salts with alkaline
agents, or during the diazotation itself when weak acids are present.
So 2-amino-1,4-dibromo-naphtalen when diazotized in presence of acetic acid and then brought to boiling leave a diazo-oxide after subsitution of a Br
atom by a -OH group (12):
2-amino-1,4-dibromo-naphtalen turns into 2-diazo-4-bromo-naphtyl-1-oxide
The same occurs with 2-amino-1-chloro-4-bromo-naphtalen. Bamberger (13) observed an identical reaction when submitting
2,4,6-tribromo-benzen-diazonium chloride to the action of an alkali, thus leading to the dibromo-diazo-oxide:
2,4,6-tribromo-benzen-diazonium chloride turns into 2-diazo-3,5-dibromo-phenoxide
----------------------------------------------------------------------------------
p 134
Orton has studied that reaction by making a comparison of various aromatic halogenoamines on their diazo-oxide speed
of formation after halogen elimination (14).
He put those into the following order, after determination of the transformation coefficient in 24 hours at ambiant temperature.
1-amino-2,4-dichlorobenzen - only traces
1-amino-2,6-dibromobenzen - 24% conversion
1-amino-2,4,6-trichlorobenzen - 63%
1-amino-2,4,6-tribromobenzen - 64%
1-amino-2,3,4,6-tetrabromobenzen - 74%
2-amino-1-chloronaphtalen - 76%
1-amino-2,4-diibromo-5-nitrobenzen - 80%
1-amino-2,4,6-tribromo-3-nitrobenzen - 93%
1-amino-2,4-dibromonaphtalen - 97%
The elimination of the halogen and the formation of the diazo-oxide happen when the pH of the medium has a value such that both hydroxide and
diazonium ions are present (15).
Many other cases have been observed and lead to patents. Like for example the diazonium derivatives of the following halogenoamines:
?-amino-2,3-dichloro-4-sulfobenzen (16),
1-amino-2,3-dichloro-4-sulfobenzen (17),
1-amino-2-chloro-3-nitro-5-sulfobenzen (18),
1-amino-2,3,6-trichloro-4-sulfobenzen (19),
2-amino-1-chloronaphtalen (20),
?-amino-2,4-dichloro-6-sulfonaphtalen (21).
Another interesting case is the transformation of the 1,5-bis-diazonium-2,6-dibromoanthraquinon bis-sulfate into a bis-diazo-oxide by treatment with
diluted sulfuric acid:
1,5-bis-diazonium-2,6-dibromoanthraquinon bis-sulfate turns into 1,5-bis-diazo-anthraquinon-2,6-dioxide
With a few halogeno-diamines was noticed that diazotation occured normaly on one of the -NH2 group and that the other one in ortho of the halogen
provided after elimination of the later a diazo-oxide. So (22):
2,6-diamino-1-chloro-4-sulfobenzen turns into 6-diazonium-2-diazo-4-sulfo-phenoxide salt
----------------------------------------------------------------------------------
p 135
2,4-diamino-1-chloro-6-sulfobenzen turns into 4-diazonium-2-diazo-6-sulfo-phenoxide salt
Elimination of the sulfo group.
Just like the groups -NO2, -OCH3 and halogens, the sulfo group in ortho position vs a -NH2 can be eliminated and replaced by -OH, with subsequent
formation of a diazo-oxide when submitting the diazonium salt to an alkaline agent.
This phenomenon is illustrated by the examples of diazonium salts of the sulfonic acids
2-amino-1,5,7-trisulfonaphtalen, 1-amino-2,4-disulfonaphtalen, 1-amino-1,5-disulfonaphtalen (there must be a mistake since
position 1 can't hold two groups at the same time) that decompose with formation of diazo-oxides (23):
8-amino-1,5-disulfonaphtalen turns into 8-diazo-5-sulfo-naphtyl-1-oxide
Into the case of diamines, we get simultaneously a diazo-oxide and a diazoic compound (diazonium).
1,3-diamino-4,6-disulfobenzen turns into 2-diazo-4-diazonium-5-sulfo-phenoxide salt
It is noteworthy that the second sulfo group is not eliminated, like the first, despite it is also positioned in ortho of a -NH2 group.(and thus that a bis-diazo-phen-dioxide is not obtained…but this may be a consequence from the impossibility to make a
meta-quinonic structure)
Migration
A good deal of cases are known where the anion of a diazonium switch its place with one of the ring groups.
Para-chlorobenzen-diazonium thiocyanate obtained by double decomposition between chlorobenzen-diazonium chloride and potassium thiocyanate, turns into
the chloride of p-thio
----------------------------------------------------------------------------------
p 136
cyanobenzen-diazonium when exposed to a mildly acidified alcoholic solution (24):
1-diazonium-4-chloro-benzen thiocyanate turns into 1-diazonium-4-thiocyano-benzen chloride
There is also a similar case of switching of Cl and Br atoms; for example, 1-diazonium-2,4,6-tribromo-benzen chloride turns into
1-diazonium-4-chloro-2,6-dibromo-benzen bromide (25):
1-diazonium-2,4,6-tribromo-benzen chloride turns into 1-diazonium-4-chloro-2,6-dibromo-benzen bromide
That transposition occurs with difficulty into an aqueous solution, but happens with more ease into an alcoholic solution, and if a brom atom is
positionned in ortho or para of the diazoic group; it must be noticed that the transposition is easier with an increasing number of bromine atoms onto
the ring; so 1-diazonium-2,4-dibromobenzen chloride salt is transformed while into alcoholic solution, but is stable when in the dry state while for
the 1-diazonium-2,4,6-tribromobenzen chloride transformation occurs even when dry.
Anion exchange does'nt happen when it goes about iodine atoms from the ring (like for 1-diazonium-2,4,6-triiodobenzen chloride), or when it goes about
fluoride anion (like for 1-diazonium-2,4,6-tribromobenzen fluoride) (26).
Once was also mention of an anion exchange involving a -NO2 group. So 2-diazonium-1-nitronaphtalen chloride turns, when in anhydrous acetic acid
media, into 2-diazonium-1-chloronaphtalen nitrite (27):
2-diazonium-1-nitronaphtalen chloride turns into 2-diazonium-1-chloronaphtalen nitrite
----------------------------------------------------------------------------------
p 137
Elimination, substitution or migration of groups into diazonium compounds. -
Bibliography*.
References 1) to 27) see picture of page 137.
Stability of diazonium salts
Stability into the solid state
Into the solid and dry state, diazonium salts are very unstable and decompose explosively by shock (1,2).
The anion has a determining effect on the unstability of the salt that increases in the following order: SO42-, < Cl-, < NO3-, ClO4-.The groups onto the ring have also a marked
effect; while halogens increase stability, -NO2 groups decrease it and, as a result, increase the tendancy to explode (3,4).
Caro and Griess did patent the use of benzen diazonium chromate as an explosive (5), but it seems that it didn't got widespread use. On
the other hand, 2-diazo-4,6-dinitrophenoxide, obtained by diazotation of picramic acid, revealed to be a powerful explosive worth getting attention
(6,7,8), and abbreviated into the United States by the letters D.D.N.P.:
picramic acid (2-amino-4,6-dinitrophenol) turns into 2-diazo-4,6-dinitrophenoxide
*See note on foot page 126 (abbreviations).
----------------------------------------------------------------------------------
p 138
That compound takes the form of a yellow crystalline powder with a true density at 25°C of 1,63. The apparent
density under a pressure of 239 kg-m² is 0,86. It is soluble into nitrobenzen from what it can be recrystallized, unsoluble into petroleum ether and
into carbon tetrachloride, weakly soluble into chloroform, into benzen, into alcohols and into ethers. Clark (9) has made a comparison of
its explosive properties vs those of mercury fulminate and of lead azide. It is less sensitive to shock as can be concluded from the following
comparison:
Minimum falling height of a 500 g weight to generate an explosion
D.D.N.P 22,5 cm
Lead azide 17,5 cm
Mercury fulminate 15 cm
Its explosive power is about three times stronger than that of lead azide. The "Sand Test" examination yielded the following results:
Charge 0,60
D.D.N.P 54
Mercury fulminate 27,5
Lead azide 21
Charge 1,00
D.D.N.P 90,6
Mercury fulminate 48,4
Lead azide 36
As initiating material, it is very close to lead azide and more potent than the fulminate.
Minimum charge to initiate explosion (detonation) of explosives
Picric acid
Mercury fulminate 0,225
D.D.N.P 0,115
Lead azide 0,12
Trinitrotoluen
Mercury fulminate 0,240
D.D.N.P 0,163
Lead azide 0,16
By the specific example of 2-diazo-4,6-dinitrophenoxide, one can see that the diazo group increases the explosive properties of dinitrophenol.
For the large amount of uses resulting from the growing use of diazonium salts as intermediary agents into the synthesis of dyes, the unstability was
a major problem that pushed to research stable or stabilized salts on what we will come back later and appart because of their importance (see p.
215).
Stability into aqueous solutions
Into solution, the decomposition reaction of diazonium salts is much slower than in the solid state.
As a result, solutions and in particular aqueous solutions, were used to study the decomposition mechanism and to compare the stability of various
diazonium salts with each other.
[Edited on 11-1-2017 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
Theoretic
National Hazard
Posts: 776
Registered: 17-6-2003
Location: London, the Land of Sun, Summer and Snow
Member Is Offline
Mood: eating the souls of dust mites
|
|
Wow great job Philou! That must have taken a lot of patience. (Although, one can get rid of many of those line breaks and make the post more compact,
and even better)
|
|
|
PHILOU Zrealone
International Hazard
Posts: 2893
Registered: 20-5-2002
Location: Brussel
Member Is Offline
Mood: Bis-diazo-dinitro-hydroquinonic
|
|
Yes took me some time.
Sadly this forum text protocol doesn't allow for the use of alignement via tabulation so I had to find another way to put tables (via insertion of
list and centering of the text). Of course I could have compacted all the text into a single block text, but the aim here was to keep the original
structure sothat one reading the initial document finds its marks directly with the translation in parallel.
If we take a deeper look at the following sentences:
------------------------------------------------------------------
"In a more general way than for the polynitro-anisidins, amines that are nitrated ortho vs the NH2 give rise to elimination of the -NO2
group during diazotation if the para position is occupied by an electro-negative group."
"Into the benzen family, 1-amino-2,4-dinitrobenzen, 1-amino-2-nitro-4-sulfo-benzen, 1-amino-2-nitro-4-arseno-benzen,
1-amino-2-nitro-4-chloro-benzen, 1-amino-2-nitro-4-chloro-5-sulfo-benzen, all lose their -NO2 group placed in ortho vs the -NH2 and lead to
the formation of the corresponding diazo-oxide by diazotation in acidic media."
"Same kind of reaction process is observed into the naphtalen family. So 2-amino-1,6-dinitro-naphtalen diazotized in presence of
concentrated sulfuric acid and then dilluted by water immediately delivers the diazo-oxide:
2-amino-1,6-dinitro-naphtalen tuns into 2-diazo-6-nitro-naphtyl-1-oxide
The same happens to 2-amino-1,8-dinitro-naphtalen and to 1-amino-2,4-dinitro-naphtalen."
------------------------------------------------------------------
We may conclude that:
1°) the presence of an electron withdrawing group or atom in para of the NH2 and the presence of a NO2 in ortho allows for elimination of the ortho
NO2. But this electron withdrawing group can be a simple chloride atom (like for the example nitrochloroanilin); or an aromatic ring activated by a
-NO2 like for the last 2-aminonaphtalen example.
2°) typical examples of dinitroanilin and dinitronaphtylamin, but also of 2,4,6-tribromoanilin allowed me to extrapolate for the case of
trinitroanilin (picramide) what would generate as said another ortho-DDNP variant (the same as the normal one but with the diazo and OH switched) thus
the one that would result from diazotation of 2-amino-3,5-dinitrophenol instead of conventional picramic acid.
3°) It seems that for NO2 elimination, basic treatment or weak acid treatment are not mandatory and that simple dillution of the sulfuric acid media
is sufficient.
4°) It seems that into concentrated H2SO4 the diazonium salts is stable and dissolved while upon dillution the diazo-oxide precipitates...and this
may be paralleled with last Nitro-genes experiments.
About other thoughts: (see picture below)
1°) It would be interesting to test diazotation for the following compounds and investigating new powerful diazo-oxide primaries
-EWG trinitromethyl in para position
--> 2-nitro-4-trinitromethylanilin
--> 2,6-dinitro-4-trinitromethylanilin
--> 2-nitro-4,6-bis-trinitromethylanilin
-EWG nitro in para position
--> 2,4-dinitro-6-trinitromethylanilin
2°) Inspired by the bis-diazo-oxide-anthraquinon case, the design of a bis-diazo-oxide compound seems very interesting because it would mean higher
density and more power... what can be interesting and desirable for initiation purpose as a primary or simply as a HE explosive:
-for benzen nucleus (with two nitro groups or one or two trinitromethyl groups)
-for naphtalen nucleus (the diazo-oxide can involve positions on the same ring side of the molecule thus a 5-membered cyclus or may involve the two
rings (like positions 1,8 and 4,5) via a 6-membered cyclus)
-for biphenyl nucleus (with two nitro groups or one or two trinitromethyl (nitroformyl) groups on each phenyl ring.
-In the case of benzen because of the unpossibility to get a bis-diazo-oxide involving a discrete meta-quinonic structure, one would have to
hypothesize that an ortho-quinonic or para-quinonic structure may hold the line.
As such one would have to investigate pyro-catechol (o-benzendiol) and hydroquinon (p-benzendiol) derivatives like:
1,2-dihydroxy-3,6-diaminobenzen (with explosophoric groups into position 4 and 4,5)
1,4-dihydroxy-2,5-diaminobenzen (with explosophoric groups into position 3 and 3,6)
1,4-dihydroxy-2,3-diaminobenzen (with explosophoric groups into position 5 and 5,6)
In this last case one may get also a competitive reaction vs the diazo-oxide formation because two NH2 are vicinals and usually this may lead to
benzo-triazole ring...what may also be interesting.
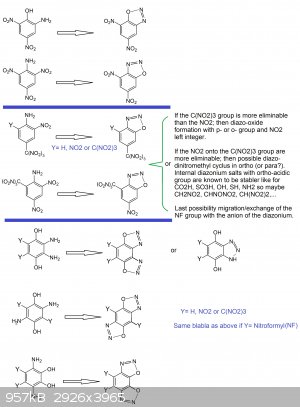
I keep for me the drawings for naphtalen and biphenyl since it involes a lot more of isomerism possible cases.
[Edited on 13-1-2017 by PHILOU Zrealone]
PH Z (PHILOU Zrealone)
"Physic is all what never works; Chemistry is all what stinks and explodes!"-"Life that deadly disease, sexually transmitted."(W.Allen)
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Thanks Philou, interesting reading material! Would have liked to try the TNA diazonium + base reaction, though I don't have anything HE related
anymore. Lots of interesting possibilities for further study, although some of the precursors to the diazophenols you describe in your last schemes
may be very difficult to obtain. I also wonder how stable any of the
bis-diazophenols would be. Adding the trinitromethyl moiety is an interesting thought, just like diazonium salts and diazophenols from coupled
molecules, biphenyls, stilbenes, maybe even some more OTC options like xanthones etc., like nitration off 2,7 diaminoxanthone or
bis(4-aminodiphenylether) For the most part, obtaining the required nitro and amino derivatives selectively from nitration and nitro reduction will
the challenging here. Maybe some other aromatic carbon rings or even heterocycles might also be able to form stable diazonium phenols so lots of
possibilities for sure!
After trying the nitration of some left over pDDNP again (Cautiously adding few mg's at a time to large amount of-20 SA), the light yellow compound is
probably the triazene from your scheme. After boiling in potassium acetate until colour change to the colour of pDDNP, the supernatant was treated
with CaCl, no precipitation, so no SO4 present. Adding extra KNO3, results in no precipiation of K-picrate. When the brown salt is reaciidified, the
light yellow colour returns. The monopotassium salt of the triazene is probably somewhat soluble, while the di-potassium salt upon basifying further
is very soluble or leads to destruction. It is interesting that nitroamidoresorcinol can actually be nitrated at low temp using KNO3/SA, still wonder
what happens here and in what order, in either case that extra hydroxyl is probably vital.
I wonder if the schffbases from your scheme may form from oxidation by SA itself, stupid... never occured to me that hot SA may be able to oxidize the
isopicramic to the quinone imine, could swear smelling some SO2, although impurities in the isopic could also be the cause here. It is very difficult
to get completely pure. Takes several crystallizations to get those beautiful golden hair-like crystals, and not all was purified to this extend.
Anyway, most puzzles solved...
[Edited on 14-1-2017 by nitro-genes]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
Ok, one more post showing the ignition of both pure crystalline p-DDNP (left) and DDNR (right) Compared to the potassium salt, the free DDNR is relatively tame. Burns somewhat quicker and leaves significantly less
residue, which is expected. One final thing that would be interesting maybe is to produce the 4-azido compound of DDNR, not sure how stable it would
be. The 4-azido 2,6 dinitrophenol is easily made by reacting 1 mole eqvt of NaN3 in 97% ethanol and leaving it stirring for several hours at room temp
until no more evolution of nitrogen is observed, perhaps this would also work for DDNR? It seems that azido phenols can also undergo ring expansion or
other internal rearangements somehow, wonder if that is the reason that 2-azido 4,6 dinitrophenol seems more stable than the 4-azido isomer, which
cannot be made from aquous solution.
Pure crystalline pDDNP was made dissolving 0.5 grams of purified isopicramic acid in 20 ml 10% w/w SA at 80 deg C. This was cooled to room temperature
and filtered to remove minor dark brown impurities. Then 0.2 grams sodium nitrite in 2 ml water was added at once and left stirring for 20 minutes.
The pDDNP is obtained as light yellow-orange plates.
DDNR was made by nitration of isopicramic acid: 15 grams 97% SA was added to 20 ml beaker. Then 1.1 grams of KNO3 was added while stirring until all
dissolved. This was cooled in an ice bath and 1 gram isopicramic acid added at once. This was left stirring for 16 hours and then slowly diluted with
water until total volume of 20 ml. This was extracted with 3X 3 ml ethylacetate at low temperature to extract the 4-diazo 2,3,6 trinitrophenol. All
ethyl acetate was left to evaporate. I originally planned to also include the ignition of 4-diazo 2,3,6 trinitrophenol in the video, but it is nearly
impossible to get dry due to solvent retaining and reactivity. Finally distilled water added and heated to 80 deg C until no fizzling could be
observed. Yield is ~100 mg of light orange-brown crystalline DDNR.
Attachment: Ignition 10 mg pDDNP (left) and DDNR (right).avi (4.6MB)
This file has been downloaded 1079 times
[Edited on 1-2-2017 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
If what you are reporting is true, then this is novel, and goes beyond what Meldola and others had reported about failure of exhaustive attempts to
further nitrate isopicramic acid. You should submit this for peer review and publication.
A good way to confirm the DDNR would be to use acetic anhydride to convert some of the paracetamol to the diacetyl prior to nitration and follow the
already documented approach to DDNR first reported by Meldola and later confirmed by Klapotke, even though he incorrectly trusted the dubious NMR data
that got the structure wrong
Anyway you could then compare the two DDNR samples gotten by different routes and see if they are identical.
The potassium and strontium salts of DDNR are likely both interesting, IIRC and barium and nickel might be also. There could also possibly be a double
nickel - potassium salt.
[Edited on 2/1/2017 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
IIRC they only tried further nitration of 2,6 dinitro 4-acetaminophenol, but I've never read how this was performed or the outcomes of different
nitration methods they tried. Whether they received an unchanged precipitate of DNAc indicating that truly no further nitration occurred or foaming
mass of NOx upon crashing in ice. I don't see how Klapotke is wrong about the structure of DDNR they published, other than perhaps nomenclature used.
I'm reasonably sure my product is DDNR, any ortho and para quinones formed would likely oxidize further in the nitration mix and both colour and
solvent retaining properties match those described for 4-diazo 2,3,6 trinitrophenol. The diazo group is obviouly present and I think it is highly
doubtful any dinitro phloroglucinol diazide could be present from hydrolysis of a putative tetranitro compound. Other than reported, it seems that
heating in distilled water also works to eliminate the 3-nitro, as opposed to a strong acetate solution. Don't think this would be a really novel or
useful reaction as most of the isopicramic is oxidized, maybe HNO3 mediated oxidation is somewhat prevented by the large amount of sulfuric and
keeping the nitrate salt to a minimum, making that a maximum ratio of NO2+/HNO3 is present in the mix. Maybe DNAc could also be hydrolysed/nitrated
further at the same time using this mix, but also here there is probably little advantage.
[Edited on 1-2-2017 by nitro-genes]
|
|
|
Rosco Bodine
Banned
Posts: 6370
Registered: 29-9-2004
Member Is Offline
Mood: analytical
|
|
I would have to review the literature again to be certain but my recollection and understanding is that isopicramic acid was the end nitration result
and no third nitro would introduce even with aggressive attempts at higher nitration using fuming HNO3.
Go back several pages to find where I identified the DDNR structural disagreement issue arising with how the Klapotke article NMR shows a 6 position
for the diazo that does not agree with classical theory for the 4 position of the diazo, and the NMR basically declares by inference a diazo
rearrangement occurred that seems very unlikely for DDNR. Both can't be true, so if the recent NMR data is correct, then a diazo rearrangement did
occur. This raises a question further if DDNR made from resorcinol is the same compound, or is it misidentified as to structure in the earlier
literature which did not detect any diazo rearrangement. It could be that there is a DDNR and an iso-DDNR similarly as there is a DDNP and an
iso-DDNP.
[Edited on 2/1/2017 by Rosco Bodine]
|
|
|
nitro-genes
International Hazard
Posts: 1048
Registered: 5-4-2005
Member Is Offline
|
|
IIRC there seemed to be quite some confusion regarding the formation of either the 2,3,5 or 2,3,6 trinitro 4-aminophenol and corresponding products
upon diazotization like you mentioned a while back. Not sure if a 2,5 dinitro 4-diazo resorcinol would exist and if this would be most likely to form
upon diazotizing the 2,3,5. In this case 2 compounds were wrongly charaterized, which seems very unlikely. If you refer to the compound as DDNR
however, then a 4 diazo would be the same as a 6 diazo right? Can imagine how challenging this must have been in a time where no FTIR/NMR etc was
available to characterize the compounds produced. In addition, many of these nitrations may produce a mix of isomers. Still wonder if this also is the
case for the nitration of paracetamol using conc SA, there is quite some fizzling during the deacetylation, which could originate both from an
N-nitroso compound or 2,3 or perhaps even 2,5 dinitroacetaminophenol. Haha, perhaps the DDNR obtained from the nitration of isopicramic is actually
from further nitration of one of the former derivatives....not likely, but who knows.
Do you have a reference for the nitration of isopicramic? The same reaction at higher temperatures using picramic acid produces mostly DDNP, for
isopicramic I would guess the same. Hmm, even 2,6 dinitro 1,4 phenylenediamine produces pDDNP upon nitration using 100% HNO3/AA, so why the sulfuric
not? Most likely it prevents premature diazotization as pDDNP doesn't nitrate further.
[Edited on 2-2-2017 by nitro-genes]
|
|
|
| Pages:
1
..
23
24
25
26
27
..
33 |
|








