| Pages:
1
..
22
23
24
25
26
..
33 |
gdflp
Super Moderator

Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Thanks. I used LaTeX to display all of the formulas and what not, the forum supports raw coding using "$$" to mark the beginning and the end of the
code. It takes a bit of time to type everything out, but it's definitely worth it for the clarity it provides. The forum uses MathJax as an
interpreter, I believe that it supports several other similar languages as well.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Quote: Originally posted by gdflp  | | Thanks. I used LaTeX to display all of the formulas and what not, the forum supports raw coding using "$$" to mark the beginning and the end of the
code. It takes a bit of time to type everything out, but it's definitely worth it for the clarity it provides. The forum uses MathJax as an
interpreter, I believe that it supports several other similar languages as well. |
$$\hat{H}\Psi=E\Psi$$
Hell... so it does! And I never knew that... Neither I think do Darkstar and annaandherdad. I use it all the time on Physics.SE and have been cursing
SM for a long time for not supporting it.
Wish I knew before: it would have made much of this thread so much clearer and more elegant... 
[Edited on 2-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Looks like great stuff gdflp. Thanks !
There are just 3 bits i'm struggling with : the first bit, the middle bit and the last bit.
So this applies to any extraction, i.e. solid extractions as well.
Is that the case where extraction solvent = starting solvent ?
| Quote: | | more extractions using lesser amounts of solvent are more efficient than fewer extractions using greater amounts of solvent |
Is this why a Soxhlet is said to be an efficient extraction tool ?
(Lots of cycles with small amounts of solvent each time)
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
I will be covering liquid-solid extractions soon, much less math involved in them. Liquid-solid extractions are when solids are directly extracted
with a solvent, whereas liquid-liquid extractions are when a solution with dissolved compounds is extracted with another solvent.
Soxhlet extractors are used for liquid-solid extractions, and there is a different reason why they are so efficient; using multiple portions of
extraction solvent is typically only done with liquid-liquid extractions. I will explain them along with liquid-solid extractions since they go hand
in hand. Would you like an explanation of rotary evaporators as well, since they fit in nicely with this topic?
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Of course !
Thanks for joining in and spending your time on this.
[Edited on 2-12-2015 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by aga |
| Quote: | | more extractions using lesser amounts of solvent are more efficient than fewer extractions using greater amounts of solvent |
Is this why a Soxhlet is said to be an efficient extraction tool ?
(Lots of cycles with small amounts of solvent each time)
|
Listen young man, I know you a bit and know that basic algebra is not beyond you. So please study that basic math in there because therein lies the
answer to your question. Math compresses many logical statements into a few lines but 'unpacking' that logic into ordinary language can be time
consuming.
/End of mild-slap-on-wrist.

If there is an object lesson to be taken away from gdflp's lecture, it's that multiple extractions using small amounts of solvent each time is MUCH
to be preferred to one (or two) extractions with large volumes of solvent.
Soxhlets, useful as they are, don't really comply with that. If I had a pound for every beginner misusing a Soxhlet I'd be rich, BTW...
[Edited on 3-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
The multiple-small-extraction message stands out for sure.
In what way do <strike>we</strike> those sad, naieve noobs misuse a soxhlet ?
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
1. Showing off the shiny glass when it's not needed.
2. Under-loading the cartridge: in the spirit of gdflp's lecture, use kind of as much of the material you want to leach something from as possible...
[Edited on 2-12-2015 by blogfast25]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Industrially, L/L extractions can also be carried out in continuous mode: a rising column of the lighter solvent with the heavier solvent falling
through it. The solute then transfers from one solvent to the other.
And fractionated distillations were in my engineering course (a million moons ago!) considered counter-current liquid-vapour extractions.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Under-loading a Soxhlet ?
Confuseled.
Stuff some cotton wool in the pipe.
Add some solvent to prevent an airlock in the syphon.
Stuff the tube with the 'stuff' up near the top of the syphon pipe.
Add some more cotton wool if the 'stuff' floats.
Is there more to it than that ?
This did not work with my rig + DCM as the cooling water was not iced.
The DCM boiled & refluxed happily Above the stuff i was trying to dissolvificate.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Not putting enough coffee in the cartridge (if you're extracting caffeine from coffee, for instance). You want a decent ratio of solid (from which to
extract) to solvent.
Two more sets of reaction mechanisms coming up but I'll give you a breather till tomorrow...
[Edited on 2-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Phew ! Many thanks !
I got :-
blogfast25's brain-melting QM info via repeated hammering on the head.
Darkstar's OC Explained via arterial injection.
gdflp's liquid sep Maths via brain stem insertion.
Could take a few moments for the Noggin to Heal a bit.
Also would like to do the phenolpthalein thing and post fotygrafs.
A good Practical would be a liquid-liquid sep (hint-hint).
Edit:
(Hint-hint-hint)
Maybe a synth that includes QM, OC and the Sep things, all in one amazing technicolour Pop.
[Edited on 2-12-2015 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
For now I suggest to follow the vid's recipe: it seems well made.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Well, i attempted the phenolpthalein synthesis and failed.
All went absolutely fine up to the point where the insoluble product was supposed to vac filter out.
The filter paper broke (a single hole broke through) and sucked the product+solution into the vac flask.
Uncertain where the product was, i added NaOH solution to go purple-hunting and found it everywhere.
One thing led to another and i ended up with 400ml of purplish basic solution,
In an effort to discover what PP is NOT soluble in, i added various reagents to samples in test tubes. I even tried salting out with
K3PO4.
An amazing result happened with ~1g K3PO4 + 2ml toluene + 2ml ethanol.
A 12 phase mixture !?!?!

Help us out here gdflp !
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Aww, that kind of sucks, especially because you failed to recover the product, post-disaster ('keep calm and carry on').
The cause is almost certainly too high acidity of your slurry: conc. HCl ruins filter paper real easy, so another filter medium or separation method
would be needed. Try repeated filtering over a PP or PE tea sieve or non-woven glass fibre. Even better here would be a high porosity glass frit
filter.
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
As it happens, i salvaged the product, albeit in the form of 400ml of milky liquid.
Hopefully, with tonight's low temperature and a bit of Time, there'll be some product to recover tomorrow or the day after.
If not, it'll get centrifuged - it will not escape.
Edit:
Guess i should post the glorious techncolour ...
[Edited on 3-12-2015 by aga]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
Will we be able to oxidise Xenon (or Krypton) with super acids soon?
Meant to be a bit of ‘Awe! Shock!’ light relief from an overdose of reaction mechanisms I realised after writing this up it might be a bit heavier
on theory than I anticipated. Oh well: bear with me anyway...
Acids and Bases Recap:
We can define an acid’s strength as its capacity to protonate bases:
$$\mathrm{HA} + \mathrm{:B} \leftrightarrow \mathrm{A^-} + \mathrm{HB^+}$$
In Brønsted–Lowry theory we use water as a base (and standard), allowing to determine an objective constant known as the K<sub>a</sub>:
$$K_a=\frac{C_AC_{H3O}}{C_{HA}}$$
An important corollary is that the protonated base BH<sup>+</sup>, in Brønsted–Lowry theory called the conjugated acid of B,
is itself an acid.
Symmetrically, the deprotonated acid A<sup>-</sup>, in Brønsted–Lowry theory called the conjugated base of HA, is itself a
base.
Most crucially the theory shows also:
The stronger the acid, the weaker its conjugated base.
The stronger the base, the weaker its conjugated acid.
Carborane Super Acids:
Super acids, with acid strengths millions of times higher than that of the most common strong mineral acid H2SO4, are nothing new and the first
carborane acid was synthesized in 1967. Since then the technology and its applications have, needless to say perhaps, come a long way.
The general structure of carborane super acids is shown below (draw that in ChemSketch!):
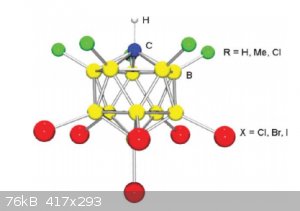
As can be seen from the possible substituents, it’s rather a big family.
Protonate anything?
In Brønsted–Lowry theory we would never consider benzene, substituted benzenes or other so-called arenes, as bases simply because the
protonating species H<sub>3</sub>O<sup>+</sup> is far from strong enough to protonate these species. Pure H2SO4 is far from
capable of protonating that stuff either: they are simply far, far too weak bases for that to happen.
But specific carboranes achieve just that and the resulting cation in the case of benzene is called benzenium -
C<sub>6</sub>H<sub>7</sub><sup>+</sup>, with the following structure:

Attentive readers might now expect the benzenium cation and the conjugated base of the carborane acid to form a Lewis base acid adduct in the usual,
here schematised way:
$$\mathrm{A^-} + \mathrm{HB^+} \to \mathrm{A-HB}$$
where a covalent bond between the Lewis acid and Lewis base forms.
But that doesn’t happen here: the beauty of carborane super acids is that their conjugated bases are extremely weak Lewis bases that
don’t form adducts, instead they form real salts: A<sup>-</sup>BH<sup>+</sup> ionic lattices. Below is the structure of a
toluenium carborate salt:
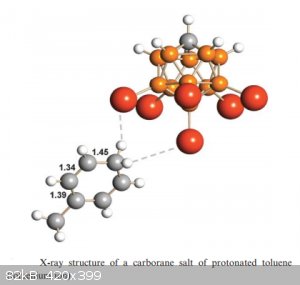
Because of the near-absolute inertness of carborate conjugated bases, carborane super acids have been referred to as “Gentle Giants” because
despite their immense strength they are non-corrosive to glass.
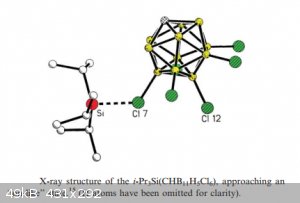
Trialkylsilylium ions:
The cation Si<sup>4+</sup> doesn’t exist because its small size and high charge surround it with a very powerful electrical field that
would snatch hydride or oxide ions immediately, in order to lower its charge and thus its energy.
Even silicon equivalents to more traditional (electron pushing stabilised) tertiary carbocations were a bit of a Holy Grail of chemistry until the
advent of carborane super acids. But at least one tri(isopropyl)silylium carborate salt has been obtained, see structure below:
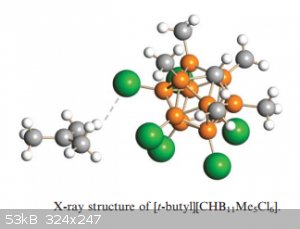
Protonating alkanes:
Even plain old alkanes, known for their general chemical inertness, aren’t safe from carborane super acids, witness below the structure of a t-butyl
carborate (which is stabilised by the electron pushing methyl groups}:
Oxonium Salts:
When we dissolve HCl in water we know that the hydrochloric acid deprotonates almost completely acc.:
$$\mathrm{HCl} +\mathrm{H_2O} \to \mathrm{Cl^-} +\mathrm{H_3O^+}$$
The solution can thus in a sense be considered as a solution of oxonium chloride. Evaporating the solvent does not however yield a salt and
this is in part due to the fact that Cl<sup>-</sup> is a very, very weak base but still too strong.
But oxonium carborates have been isolated, like the one below:
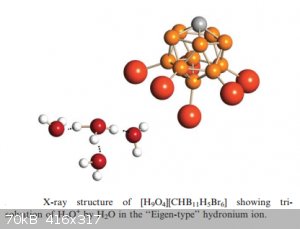
Less well known is that besides H<sub>3</sub>O<sup>+</sup>, there’s a whole zoo of similar (but less abundant) oxonium ions
out there, like: $$\mathrm{ H_5O_2^+},\:\mathrm{ H_7O_3^+},\:\mathrm{ H_9O_4^+} ...$$
Oxidising Xe (or Kr) with Carborane Super Acids?
A traditional method of oxidising metals is by means of Brønsted–Lowry acids, here for a generic bivalent metal:
$$\mathrm{M} + 2\:\mathrm{ H_3O^+} \to \mathrm{M^{2+}} + \mathrm{H_2} + 2\:\mathrm{H_2O}$$
... for metals with an SRP < 0 V.
Is it possible that the following reaction will be possible with future super acids?
$$\mathrm{Xe} + 2\:\mathrm{HCB} \to \mathrm{Xe^{2+}} + 2\:\mathrm{CB^-} +\mathrm{H_2}$$
Source.
[Edited on 4-12-2015 by blogfast25]
|
|
|
aga
Forum Drunkard
Posts: 7030
Registered: 25-3-2014
Member Is Offline
|
|
Phenolpthalein Synthesis
As per Nile Red's process https://www.youtube.com/watch?v=kBo5UnNRodA
2g Phenol
1.5g Pthalic Anhydride
1ml of 96 w% H2SO4
were added to a 25ml RBF with a magentic stir bar.
Immediately some orange colour was seen.
The RBF was suspended in a mineral oil bath (SS dog bowl + baby oil) on a hotplate/stirrer.

The stirrer and heating were switched on and the bath allowed to heat to 150 C.
The oil begain to smoke at 150 C so the temperature was reduced to 130 C. Steam was still seen to be escaping at 130 C.
Any future attempt at this method will use a small beaker rather than an RBF and an extraction fan to more efficiently remove the steam from the
reaction water.
After 1 hour the mixture took on a more Orange colour.

The RBF was removed and swirled to re-incorporate the pthalic anhydride stuck higher up the flask.
At 2 hours the mixture was a very dark purple mobile liquid.

The white crystal growth around the neck of the RBF became very noticeable, with crystals around 10cm long protruding down from the glass neck. These
were scraped with a glass rod back into the reaction mixture.
On Cooling the reaction mixture became a thick tar, trapping the stirbar.
Adding 10ml distilled water + 10ml dichloromethane (DCM) did nothing to dissolve the tar.
(In future experiments the 10ml water would be added to the hot mixture. The larger volume of water should (hopefully) not flash-boil yet keep the
product mobile.)
Freeing the stirbar using a glass rod was difficult, however once free, and turned to 100% stir speed the tar began to dissolve.
By angling the RBF over the more stubborn tar, most of the tar was dissolved after 15 minutes.
When stirring was stopped, the mixture formed two layers within 4 minutes.

The RBF contents were transferred to a 100ml separatory funnel, then ~2ml of DCM were used to wash the residues from the RBF into the sep funnel..
The layers separated within 5 minutes.
The Lower Layer was drained into a 100ml beaker.
10ml of DCM was added to the sep funnel, which was capped, then shaken and vented 5 times.
After 5 minutes the layers were separated, and the Lower Layer was drained into the same beaker as the previous lower layer.
The Upper layer was drained into a beaker for later disposal.
After washing the sep funnel with DIW twice, the combined Lower Layers were added back to the sep funnel, and 5ml of 2[M] NaOH solution was added.
This was capped, then shaken and vented 10 times.
It went purple, and the layers separated.

The Lower layer was drained into a 100ml beaker for later disposal.
The product-rich Upper layer was drained into a fresh 100ml beaker, then the sep funnel was washed with a small amount of DIW to flush the remainder
into the same beaker.
The collected product was poured into 100ml 2 [M] HCl solution.

At this point the product was vac filtered, and the filter paper devolped a hole, creating a whole new world, including 12 phase mixtures.
(it didn't work)
The Product simply WILL be recovered somehow.
Resistance is futile.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
aga:
Looks well done except for the filtering mishap, which is at 2 M HCl is not really explained by acidity attacking the filter. Perhaps it was
a faulty filter or vacuum was too high?
BTW, if you actually want to use your phenolphthalein (as a pH indicator) you'll have to wash it: ice cold DIW, a few small aliquots of it should do
it, to rinse off the acid. Or some solvent in which it is insoluble but that is miscible with water.
[Edited on 4-12-2015 by blogfast25]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
I agree with blogfast, as long as you're using some sort of laboratory filter paper, it shouldn't break due to 2M HCl. Typically, you only run into
issues with more concentrated acids, such as the solution from quenching a nitration. What are you using as a vacuum source? If you're using a
rotary pump, I would try introducing a small air bleed into the system to reduce the vacuum; since even a vacuum around 150 torr is plenty for
filtrations with reasonably coarse precipitates such as this. Acidic solutions at these concentrations may weaken the filter paper, and the pressure
from a strong pump might cause it to tear. Worst case, you could try a gravity filtration, it will just be many painful minutes/hours longer.
As for the rest of the rest of the procedure, nice job! The initial dissolution of the crude phenolphthalein is an example of a liquid-solid
extraction, and the migration of the phenolphthalein from the dichloromethane layer into the alkaline aqueous layer is an example of a liquid-liquid
extraction, albeit a slightly special case. Both are going to be covered very soon.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
I also suggest to deep-clean your glassware used: my experience with PhPht is that it sticks to anything quite enthusiastically, especially glass. Use
warm, alkaline water, so you can detect the last traces of it and scrub them off.
And no OC synth lab report is complete without a yield determination. 
[Edited on 4-12-2015 by blogfast25]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
Small traces also quite happily remain on stir bars. More than once I've dropped a seemingly clean stir bar into an alkaline reaction mixture, only
to have the entire thing turn bright pink. It's quite aggravating, it pissed me off enough that I bought a few extras which I keep separate and use
solely for pH sensitive dyes like phenolphthalein.
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by gdflp | | Small traces also quite happily remain on stir bars. More than once I've dropped a seemingly clean stir bar into an alkaline reaction mixture, only
to have the entire thing turn bright pink. It's quite aggravating, it pissed me off enough that I bought a few extras which I keep separate and use
solely for pH sensitive dyes like phenolphthalein. |
Yeah, it's really stubborn, isn't it? You can have some real mystifying and very pink surprises if you don't remember/understand PhPht was
involved...
[Edited on 4-12-2015 by blogfast25]
|
|
|
gdflp
Super Moderator
Posts: 1320
Registered: 14-2-2014
Location: NY, USA
Member Is Offline
Mood: Staring at code
|
|
<b>Liquid-Liquid Extractions : pH Manipulation</b>
Liquid-liquid extractions are a powerful tool for separating organic compounds, but there are several tricks to increase the number of situations in
which they can be used. One of these tricks is modifying the pH of the aqueous portion of an extraction to manipulate the solubility of one of the
components. By either raising or lowering the pH of the solution, the aqueous and organic solubility of compounds can be changed quite drastically,
for example a saturated aqueous solution of triethylamine at 25°C is 0.5M whereas a solution of triethylammonium chloride, the salt formed when
triethylamine is precisely neutralized with hydrochloric acid, is 10.5M. The two common classes of organic compounds which experience these drastic
solubility changes are amines, which become soluble in acidic solutions, and carboxylic acids, which become soluble in basic solutions. There are
other compounds with low pK<sub>a</sub>s, but these are the most commonly encountered.
<b>Amines and Carboxylic Acids</b>
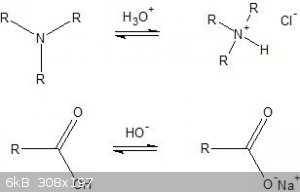
As you can see by the above diagram, in acidic solutions, amines become significantly more water soluble due to the ionic bond present in the new
compound, a salt. Likewise, in basic solutions, carboxylic acids become significantly more water soluble as their corresponding ionic salt. This
technique can be used to separate amines and carboxylic acids from neutral organic compounds. Separation is accomplished by first creating a solution
of an amine or carboxylic acid and a neutral organic compound in an organic solvent such as diethyl ether or dichloromethane, then washing with either
a dilute(~1 - 2M) solution of an acid, typically hydrochloric acid, or a base, typically sodium hydroxide, depending on the substrate.
An example of this is the phenolphthalein synthesis which you did; since phenolphthalein contains acidic protons, it can be treated as a carboxylic
acid with respect to this method. The dichloromethane solution of phenolphthalein containing various byproducts was washed with 2M sodium hydroxide,
which allowed the phenolphthalein to migrate from the organic layer to the aqueous layer, leaving behind impurities. The solution was then
reacidified to reduce the water solubility of the phenolphthalein and allow it to be recovered, as is typical with these types of extractions.
[Edited on 12-4-2015 by gdflp]
|
|
|
blogfast25
International Hazard
Posts: 10562
Registered: 3-2-2008
Location: Neverland
Member Is Offline
Mood: No Mood
|
|
| Quote: Originally posted by gdflp |
3An example of this is the phenolphthalein synthesis which you did; since phenolphthalein contains acidic protons, it can be treated as a carboxylic
acid with respect to this method.
|
Someone will have to explain that to aga, using Brønsted–Lowry (BL) theory with regards to pH indicators, their 'pH-chromism' and that they are
invariably weak acids... to get that message across. Unless gdflp wants to oblige now, I will do it tomorrow.
Very nice post, BTW. 
[Edited on 4-12-2015 by blogfast25]
|
|
|
| Pages:
1
..
22
23
24
25
26
..
33 |








