| Pages:
1
2
3 |
Filemon
Hazard to Others

Posts: 126
Registered: 26-4-2006
Member Is Offline
Mood: No Mood
|
|
It is also possible to make the condensation nitroalcohol with KOH, increasing the pH at 8 or 10. It is not necessary agitation because it is
dissolved.
http://www.orgsyn.org/orgsyn/pdfs/CV5P0833.pdf
|
|
|
smuv
National Hazard
Posts: 842
Registered: 2-5-2007
Member Is Offline
Mood: Jingoistic
|
|
A reference for the the rambles of niggaknow from orgsynth . This is a reaction I was not aware of and is pretty interesting.
@niggaknow, if you want people to take you seriously try to express yourself more eloquently.
P.S. Why did you even bother editing that post twice?
|
|
|
niggaknow
Harmless
Posts: 6
Registered: 3-2-2008
Member Is Offline
Mood: No Mood
|
|
Di bee da bomb!
2243295
http://www.pat2pdf.org
|
|
|
wa gwan
Harmless
Posts: 37
Registered: 15-4-2005
Member Is Offline
Mood: No Mood
|
|
I vaguely remember reading something about reacting alkanophenones with N-bromoalkylamines. So for example N-bromoethylamine would brominate and
ethylaminate propiophenone all in one pot without all the joys the two step reaction has to offer.
Anyone familiar with this?
It could be a patent or journal article but I've got the feeling it was in german if that helps.
[Edited on by wa gwan]
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
You could have tried using the search engine:
https://sciencemadness.org/talk/viewthread.php?tid=3963
PS: You can not use N-bromoethylamine, only the N-bromo secondary amines.
|
|
|
Drunkguy
Hazard to Others
Posts: 172
Registered: 23-12-2005
Member Is Offline
Mood: somewhat pissed.
|
|
It's not even from a patent, it was a literature entry.
| Quote: | Originally posted by stoichiometric_steve
you should try the reaction as described in the patent. there is very, VERY little conversion to the nitroalcohol under the conditions the authors
"used" (why does it all work so beautifully when typed up into a patent?). all you get is a thick, viscous mass of triethylamine - ethylnitronate.
this is supposed to react with benzaldehyde at -10°C, but surely not in a matter of a few hours - not without solvent and vigorous stirring! also,
there is no reasonable workup given.
is it even possible to distill the nitroalcohol without the risk of dehydration?
under these aspects, the propiophenone route looks much, much better, especially the "one-pot" route with DCM/NMP. |
Attachment: Henry Reaction.pdf (58kB)
This file has been downloaded 2546 times
|
|
|
stoichiometric_steve
National Hazard
Posts: 827
Registered: 14-12-2005
Member Is Offline
Mood: satyric
|
|
| Quote: | Originally posted by Drunkguy
It's not even from a patent, it was a literature entry. |
that reference looks even more promising. if i ever have the opportunity to try it out, i will report it here. although i have my doubts with H2O as
solvent...
|
|
|
wa gwan
Harmless
Posts: 37
Registered: 15-4-2005
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Nicodem
You could have tried using the search engine |
I could but I wasn't that interested. If I had been I'd have made of point of remembering it the first time I read it. 
|
|
|
unome
Hazard to Others
Posts: 134
Registered: 17-10-2009
Member Is Offline
Mood: No Mood
|
|
Propiophenone can be got to via known routes (from benzoic acid and propionic acid by the procedure of Granito & Schultz – attached), there are
ref’s from Psychokitty (mainly, although many worked on it, for example, here) on how to brominate propiophenone using Cupric Bromide to a-bromopropiophenone which could then be used in another known route (that of Hyde, Browning & Adams, also attached), ie. by
adding the a-bromopropiophenone to an 30% alcoholic solution of methylamine to give Methcathinone in fairly ordinary yields (57% cited), using Adam's
Catalyst, appropriately enough, they reduced that to racemic ephedrine/pseudoephedrine...
[img]https://www.sciencemadness.org/whisper/files.php?pid=165102&aid=8931[/img]
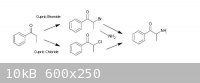
Given the difficulty involved in finding bromine in any useable quantity, it became necessary to examine whether any similar routes could be used with
chlorine. There is a series of journal articles of interest (one in particular, which is attached - Kosower, et al), which claims good yields of
a-chloropropiophenone via the Cupric Chloride chlorination of propiophenone. I am assuming that this could be utilized similarly to the a-bromo derivative to give Methcathinone
as well. I have also attached one of the MPV Reduction of Methcathinone papers (the one with the note on the ephedrine series, all that says is that
one shouldn’t try and use the bromohydrin (ie. the reduced product with methylamine – ie. use a modified version of that used by Hyde, et al). To
top all that off, I have also attached a paper in which Mandelate esters are catalytically reduced to the phenylacetic acid by refluxing with excess
Raney Nickel (wonder if precipitated Nickel/Alumina or Urushibara type would work?) in ethanol.
PS The only person I've seen who has claimed any real success with the production of propiophenone from Benzoic & Propionic acid + Iron salts, is
Organikum. Perhaps those who are interested might care to look through the Benzene from Benzoic acid threads for his (1) reaction vessel (2) procedure and (3) what he says vis-a-vis black iron oxide pigments. He was (maybe still is) an obnoxious prick at times, but he did KNOW his stuff
Attachment: Bromination.Cupric.Bromide.pdf (391kB)
This file has been downloaded 1744 times
Attachment: Chlorination.Cupric.Chloride.pdf (556kB)
This file has been downloaded 1708 times
Attachment: CTH.RaNi.EtOH.EthylMandelate.pdf (413kB)
This file has been downloaded 1491 times
Attachment: MPV.Bromoketones.NB.Ephedrine.Series.pdf (571kB)
This file has been downloaded 2053 times
Attachment: Synthetic.Homologs.of.Ephedrine.Hyde.etal..pdf (400kB)
This file has been downloaded 1616 times
Attachment: Decarboxylation.StudiesII.Prep.Alkyl.Phenyl.Ketones.Granito.Schultz.pdf (422kB)
This file has been downloaded 1783 times
[Edited on 30-10-2009 by unome]
|
|
|
unome
Hazard to Others
Posts: 134
Registered: 17-10-2009
Member Is Offline
Mood: No Mood
|
|
Here is a procedure in which the authors (Patrick, McBee & Hass) prepare the relevant benzedrine derivative by displacement of
1-phenyl-2-chloropropane with either ammonia/methylamine, which is the pretty much the final link in the chain
Attachment: amination.phenyl-2-chloropropane.pdf (359kB)
This file has been downloaded 2242 times
|
|
|
VestriDeus
Harmless
Posts: 16
Registered: 15-11-2009
Member Is Offline
Mood: No Mood
|
|
Isn't ephedrine like a schedule II controlled substance??
|
|
|
entropy51
Gone, but not forgotten
Posts: 1612
Registered: 30-5-2009
Member Is Offline
Mood: Fissile
|
|
No, but sales are tightly regulated and monitored
closely.
|
|
|
unome
Hazard to Others
Posts: 134
Registered: 17-10-2009
Member Is Offline
Mood: No Mood
|
|
I've been thinking about something, we've all seen the papers where students are shown how to make Bupropion? Well, they use Br2 in CH2Cl2 to brominate propiophenone in no time flat (and virtually 100% yield), remove the CH2Cl2 (distillation
@40'C), thus isolating the nasty ketone without having to come into contact with it.
They then alkylate t-butylamine in NMP which is a good solvent, water miscible, which simplifies workup as they only take the organic layer once they
base out the alkaloid.
What about a modification of that for Methcathinone? Methcathinone has been made before now by dropping a-bromopropiophenone into an ice cold
alcoholic (read absolute ethanol or thereabouts) solution of methylamine...
| Quote: | Taken from:
SYNTHETIC HOMOLOGS OF d,l-EPHEDRINE
J. F. Hyde, E. Browning and Roger Adams
J. Am. Chem. Soc., 1928, 50 (8), pp 2287–2292
DOI: 10.1021/ja01395a032
Page 4/6 (2290):
Reaction of the Bromo Ketones with Methylamine.
One-tenth of a mole of the bromo ketone was added drop-wise with vigorous stirring to 0.25 mole of methylamine (in the form of a 30% solution in
absolute alcohol) over the required period of time (one hour for bromopropiophenone, seventeen to eighteen hours for a-bromobutyrophenone, twenty-four
hours for a-bromovalerophenone). The reaction flask was immersed in ice water during the reaction and stirring was continued for one-half to
three-quarters of an hour after the addition of the bromo compound. Cold, concd. hydrochloric acid was then added very slowly along with some finely
cracked ice until the mixture was acidic. If it became warm the product turned very dark in color and a larger proportion of tar was produced. At this
point the reaction mixture was orange or red due to the presence of some bromo ketone that had not reacted and to the formation of certain tarry
by-products. These were extracted with ether from the water layer and the bromo ketone recovered. The water layer was evaporated to dryness in vacuo,
treated with a little chloroform and evaporated to dryness again to assist in removing the moisture from the rather hard mass. After standing in a
vacuum desiccator for a day, the residue was extracted several times with fresh portions of chloroform and each time the insoluble crystals of
methylamine hydrochloride were filtered. The chloroform solution was then evaporated until it was very concentrated, and acetone was added to cause
the crystallization of the amino ketone hydrochloride. Recrystallization was carried out by dissolving in a small amount of alcohol, filtering, and
adding about twice the volume of acetone in small portions. |
Personally, albeit hypothetically, I'd have to suggest utilizing the isolation procedure outlined in the Bupropion synthesis... The solubility of
Methylamine base in water is 108g/100mL, whereas the solubility of Methcathinone base in water is virtually nil.
I'd suggest adding a non-polar solvent, toluene/xylene/etc. then removing the volatile(s), ie. any remaining methylamine base and the ethanol.
No nasty, hard to get chemicals (saving methylamine, propiophenone & bromine of course).
PS There are MANY, MANY articles that state categorically that hydrogenation of ephedrone with 10% Pd/C @ 55psig (~4Atm IIRC) gives desoxyephedrine
directly.
[Edited on 24-12-2009 by unome]
Attachment: Hyde.Browning.Adams.Synthetic.Homologs.of.Ephedrine.Hyde.etal..pdf (400kB)
This file has been downloaded 1595 times
[Edited on 24-12-2009 by unome]
[Edited on 24-12-2009 by unome]
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
May NaBH4 or LiAlH4 use for make ephedrine from ehedrone?(replace for hydrogenation step by Raney Nickel)
NaBH4 and LiAlH4 is reducing agent and convert Ketones to Alcohols
[Edited on 14-3-2010 by Waffles SS]
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
If I'm not mistaken, NaBH4's specialty is converting carbonyl oxygens to alcohols. LiAlH4, not so much.
|
|
|
NitratedKittens
Hazard to Others
Posts: 131
Registered: 13-4-2015
Location: In the basket with all the other kittens
Member Is Offline
Mood: Carbonated
|
|
Just how is this not in detritus right now?
Basket of kittens for you ........BOOM
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Better question: why did Melgar, who usually knows what he's doing, bump a thread that was last replied to in 2010?
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Too many tabs open and I forgot which one was which. Noticed someone say something that didn't seem accurate, and did a quick reply. Sorry, my bad.
On the other hand, this is like the first time anyone here has said anything nice about me in years! Everyone is normally such pricks here, including
me, come to think of it.
|
|
|
Waffles SS
Fighter
Posts: 998
Registered: 7-12-2009
Member Is Offline
|
|
I am sorry for my stupid 7 years ago question.
[Edited on 31-3-2017 by Waffles SS]
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
It's okay Waffles, you've come a long way since then!
Incidentally, I managed to get my hands on some propiophenone, and am looking for interesting reactions that use it. The one referenced earlier in
this thread sounds really interesting, which is why I had it open in the first place:
http://www.orgsyn.org/demo.aspx?prep=cv2p0363
However, it would be nice to know if there's any possible alternative to HCl gas. If I'm going to screw around with HCl gas that's not incorporated
into the end product, I may as well make HBr gas, so I can collect the vapors in water and save them for later. My thought is that the yields would
improve slightly with HBr, but I could very easily be wrong.
Also, I seem to be able to store alkyl nitrites indefinitely, by adding some sodium carbonate to the containers, so how important is it really, that
the nitrites be freshly prepared? Is it just so the stoichometry comes out right?
[Edited on 3/31/17 by Melgar]
|
|
|
karlos³
International Hazard
Posts: 1520
Registered: 10-1-2011
Location: yes!
Member Is Offline
Mood: oxazolidinic 8)
|
|
A nice alternative to HCl gas?
I´ve got one for you, tested and tried!
Look here, that was my reference to use this reaction on propiophenone, which worked very good on it.
http://chemistry.mdma.ch/hiveboard/novel/000492480.html'
Try it! Yield is at least as good as using acidic catalyst!
Edit: Of course the nitro ester should be distilled freshly in any case.
[Edited on 31-3-2017 by karlos³]
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Wait... this is an isonitroso compound, not a nitro anything. At the end, the one guy says it's reducing a nitroalkene to an amine, which isn't
actually true, since it's just a ketoxime, which is what "isonitroso" is an old term for. Oximes, being imines of hydroxylamine, which are apparently
far more stable than ordinary imines.
It uses sodium ethoxide though, which basically necessitates that you have metallic sodium. (Which I do, but when coming up with novel synthetic
routes, the more OTC the better, IMO) I wonder if aluminum ethoxide would suffice?
| Quote: | | Edit: Of course the nitro ester should be distilled freshly in any case. |
Nitro esters... you mean like nitroglycerin? Or ethyl nitrate?
[Edited on 3/31/17 by Melgar]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: | | It uses sodium ethoxide though, which basically necessitates that you have metallic sodium. |
To post this on the forum that made famous the OTC synthesis of sodium ethoxide?
http://www.sciencemadness.org/talk/viewthread.php?tid=2656
|
|
|
Melgar
Anti-Spam Agent
Posts: 2004
Registered: 23-2-2010
Location: Connecticut
Member Is Offline
Mood: Estrified
|
|
Hence, the reason I qualify all my statements with words like "basically", "probably", "likely", and "virtually". There's no such thing as 100%
certainty in science!
It seems to work based on the fact that a saturated NaOH solution is heavier than a sodium alkoxide solution, and sodium ethoxide is much more soluble
in ethanol than sodium hydroxide is. This makes me wonder if you could do it as a batch, by putting quicklime (calcium oxide) at the bottom. Or
stirring anhydrous ethanol and sodium hydroxide with a quicklime suspension. Or if it'd be possible to have an electrolysis cell going with sodium
hydroxide in alcohol? I never really thought much about routes to alkali alkoxides, just because I've rarely had much use for one.
edit: Ok, so I set up an experiment with potassium hydroxide in methanol. That was the only alcohol I had that was more than 95% pure, other than
n-butanol. I don't quite know why I opted for potassium. There are definitely bubbles at the cathode, and they're very fine. Over time, if this is
generating an alkoxide, current should increase, as the concentration of the alkoxide makes it more conductive, right? And if it's not, the current
should decrease, since then it'd just be using residual water to generate the hydrogen, which would gradually get used up.
edit2: I'd still be curious if aluminum ethoxide would work for a reaction like this.
[Edited on 4/1/17 by Melgar]
|
|
|
clearly_not_atara
International Hazard
Posts: 2787
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
| Quote: | | Ok, so I set up an experiment with potassium hydroxide in methanol. That was the only alcohol I had that was more than 95% pure, other than n-butanol.
I don't quite know why I opted for potassium. There are definitely bubbles at the cathode, and they're very fine. Over time, if this is generating an
alkoxide, current should increase, as the concentration of the alkoxide makes it more conductive, right? And if it's not, the current should decrease,
since then it'd just be using residual water to generate the hydrogen, which would gradually get used up. |
Methanol is unique among aliphatic alcohols in that it's more acidic than water (pKa 15.5 vs 15.7) so methoxide is the predominating species in any
mixture of KOH and methanol. However IIRC it is not practical to dehydrate methanol with KOH because the solubility is so high, owing to methanol's
acidity.
I believe such solutions can also be dehydrated over K2CO3.
|
|
|
| Pages:
1
2
3 |










