| Pages:
1
..
13
14
15
16
17 |
Lionel Spanner
Hazard to Others

Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
A major breakthrough!
One of the biggest problems in producing nitrite by reduction of nitrate is its tendency to react with oxygen at the reaction temperature. The
solution? Remove oxygen.
I recently got an argon cylinder over the counter from a local welding supply store (Machine Mart) and decided to retry the sodium sulphide reduction
method described in Morgan, in the same manner as the molten lead method (reductant added to molten nitrate in portions), while sparging the flask
with argon and keeping oxygen out.
It only went and bloody well worked!
I'll need to reproduce and refine the method before providing a full write-up, but this is definitely a viable way to produce nitrites at a small
scale. Get in!
|
|
|
clearly_not_atara
International Hazard
Posts: 2826
Registered: 3-11-2013
Member Is Offline
Mood: Big
|
|
Chalk up another victory to the inert atmosphere! First eugenol demethylation, now nitrite.
Lovely choice of reducing agents we have. Sodium sulfide or lead. Any word on nickel carbonyl? 
But seriously, nice work!
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
To be fair, sodium sulphide is easily obtainable from photography suppliers (it's the traditional reagent used for sepia toning), and you only need 1
mole of sulphide per 4 moles of nitrate. Plus, technical grade sodium sulphide is hydrated and is a liquid at the temperature the reaction is carried
out, so the reaction mixture is uniform, and you don't have the problem of uneven mixing that you'd get with a solid/liquid or solid/solid mixture.
The process described in Morgan was carried out in iron pans, with no mention of an inert atmosphere, which is a recipe for failure (and an awful lot
of ammonia.) The chemistry itself was sound, but the process was pretty bad.
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
I couldn't reproduce this method.
In my initial attempt, I stopped when addition of sulphide started producing small deflagrations at the surface of the molten mixture, likely due to
formation of elemental sulphur - this started happening when 60% of the sulphide had been added. However, when I crystallised out nitrite, it was only
60% pure, suggesting an incomplete reaction had taken place. So the second time, I added all of the sulphide.
On dissolving the solidified reaction mixture, it appears the nitrite had been destroyed in the reaction, most likely due to sulphide reacting with
nitrite, forming elemental sulphur and ammonia; as no ammonia vapours were evident during the reaction, it was most likely captured as ammonium
sulphate. This would explain why although a highly soluble hygroscopic substance was produced on concentrating the mixture to near-dryness under
vacuum, it did not look like either sodium nitrate or nitrite (smaller crystals), and the pH of a solution was too low for it to be either nitrite or
nitrate (about 5).
Sod the shipping costs, going forward I'm just buying it from Poland.
|
|
|
fx-991ex
Hazard to Others
Posts: 109
Registered: 20-5-2023
Member Is Offline
|
|
In the first post he do NaHSO3 + CaCl2 and then heat the resulting Ca(HSO3)2 to decompose to CaSO3 + H2SO3.
I was thinking.
Lets start with sodium metabisulfite.
Na2S2O5 + H2O = NaHSO3
Then
NaHSO3 + Ca(OH)2 = CaSO3 + NaOH + H2O
Can this be done?(i know weak base displace stronger base, not the opposite, but the low solubility of CaSO3 will drive it forward?)
or (this should definitely work)
NaHSO3 + NaOH = Na2SO3 + H2O
followed by
Na2SO3 + CaCl2 = CaSO3 + NaCl
Also about the thermal reduction of NaNO3 + CaSO3, i plan to do it in a crucible(porcelain) with bunsen burner.
Do i heat it till the nitrate/nitrite melt?, is it gonna be hard to tell when it reached completion? or theres some color change?.
[Edited on 19-6-2023 by fx-991ex]
[Edited on 19-6-2023 by fx-991ex]
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Quote: Originally posted by fx-991ex  | In the first post he do NaHSO3 + CaCl2 and then heat the resulting Ca(HSO3)2 to decompose to CaSO3 + H2SO3.
I was thinking.
Lets start with sodium metabisulfite.
Na2S2O5 + H2O = NaHSO3
Then
NaHSO3 + Ca(OH)2 = CaSO3 + NaOH + H2O
Can this be done?(i know weak base displace stronger base, not the opposite, but the low solubility of CaSO3 will drive it forward?)
or (this should definitely work)
NaHSO3 + NaOH = Na2SO3 + H2O
followed by
Na2SO3 + CaCl2 = CaSO3 + NaCl
Also about the thermal reduction of NaNO3 + CaSO3, i plan to do it in a crucible(porcelain) with bunsen burner.
Do i heat it till the nitrate/nitrite melt?, is it gonna be hard to tell when it reached completion? or theres some color change?.
[Edited on 19-6-2023 by fx-991ex]
[Edited on 19-6-2023 by fx-991ex] |
My only comment is that you will need to work with an inert atmosphere, as nitrite salts are rapidly oxidised to nitrate by oxygen in their molten
state.
Good luck!
|
|
|
fx-991ex
Hazard to Others
Posts: 109
Registered: 20-5-2023
Member Is Offline
|
|
| Quote: Originally posted by Lionel Spanner | | Quote: Originally posted by fx-991ex | In the first post he do NaHSO3 + CaCl2 and then heat the resulting Ca(HSO3)2 to decompose to CaSO3 + H2SO3.
I was thinking.
Lets start with sodium metabisulfite.
Na2S2O5 + H2O = NaHSO3
Then
NaHSO3 + Ca(OH)2 = CaSO3 + NaOH + H2O
Can this be done?(i know weak base displace stronger base, not the opposite, but the low solubility of CaSO3 will drive it forward?)
or (this should definitely work)
NaHSO3 + NaOH = Na2SO3 + H2O
followed by
Na2SO3 + CaCl2 = CaSO3 + NaCl
Also about the thermal reduction of NaNO3 + CaSO3, i plan to do it in a crucible(porcelain) with bunsen burner.
Do i heat it till the nitrate/nitrite melt?, is it gonna be hard to tell when it reached completion? or theres some color change?.
[Edited on 19-6-2023 by fx-991ex]
[Edited on 19-6-2023 by fx-991ex] |
My only comment is that you will need to work with an inert atmosphere, as nitrite salts are rapidly oxidised to nitrate by oxygen in their molten
state.
Good luck! |
Apparently it can be done without melting the mixture (230C-300C)
Heres a video where it seem to be working well: https://www.youtube.com/watch?v=5BLPoE6Y-ns
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
If you can actually get it to work, that'd be one hell of an achievement.
As I said: good luck!
|
|
|
Sir_Gawain
Hazard to Others
Posts: 490
Registered: 12-10-2022
Location: [REDACTED]
Member Is Offline
Mood: Still in 2022
|
|
Would it work better with potassium nitrate? KNO2 is 10 times more soluble than KNO3.
“Alchemy is trying to turn things yellow; chemistry is trying to avoid things turning yellow.” -Tom deP.
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Since reductive pathways to nitrite generally require high temperatures and produce poor purity products, or result in over-reduction to ammonia, it
may be worth taking a cue from the industrial synthesis, namely the reaction between nitric oxide and caustic soda, which doesn't involve any
oxidation or reduction at all.
Now nitric oxide itself is hard to come by for amateurs, but here's an idea for an indirect route: prepare nitrosyl sulphuric acid, dissolve it in
sulphuric acid, cool the solution to near 0 °C, then add it (slowly and carefully!) to an equally cold hydroxide solution, under an inert atmosphere,
to produce a mixture of nitrite and sulphate, that can easily be separated due to the large difference in solubility.
Brauer claims direct addition of water to nitrosyl sulphuric acid produces dinitrogen trioxide, a direct precursor to inorganic and organic nitrites,
but this is probably only true at temperatures well below zero.
Nitrosyl sulphuric acid is not trivial to make, as it requires sulphur dioxide to be bubbled through cold fuming nitric acid, but as a non-volatile
solid with a melting point of 70 °C it is easier to handle than other simple inorganic nitrosyl compounds, which are highly toxic gases or
low-boiling liquids.
[Edited on 2-8-2023 by Lionel Spanner]
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Here's what may be a very interesting and relevant paper, describing selective production of nitrite or nitrate by electrochemical oxidation of
ammonia with a copper electrode.
https://chemistry-europe.onlinelibrary.wiley.com/doi/epdf/10...
Unfortunately Sci-Hub hasn't indexed it, and I'm not in a position to drop the Britbongland equivalent of $49 just to download it, especially if it
turns out to be hot garbage. Would anyone who has access to the Wiley online library be able to share it? Many thanks!
|
|
|
Parakeet
Hazard to Self
Posts: 75
Registered: 22-12-2022
Location: Japan
Member Is Offline
Mood: V (V)
|
|
Here it is.
Haven't read everything yet, but the procedure looks amateur friendly.
Attachment: ChemSusChem - 2021 - Johnston.pdf (714kB)
This file has been downloaded 362 times
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
Fantastic, thank you very much!
|
|
|
unionised
International Hazard
Posts: 5135
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Thanks but...
"Large-scale nitrite production (NaNO2) is achieved by bubbling gaseous N2O and NO through a solution of NaOH and Na2CO3".
Really?
|
|
|
Parakeet
Hazard to Self
Posts: 75
Registered: 22-12-2022
Location: Japan
Member Is Offline
Mood: V (V)
|
|
| Quote: Originally posted by unionised |
Thanks but...
"Large-scale nitrite production (NaNO2) is achieved by bubbling gaseous N2O and NO through a solution of NaOH and Na2CO3".
Really? |
Yeah. I've also read the third reference. It should be NO2.
|
|
|
fx-991ex
Hazard to Others
Posts: 109
Registered: 20-5-2023
Member Is Offline
|
|
In water it is, but in Alcohol the sodium nitrite salt is more soluble, and since alcohol is easier to remove thats why i went with the sodium salt
instead of the potassium one.
I did try the reaction(didnt extract with alcohol yet)
When i mixed the sulfite and nitrate it was a bit endothermic, so it seem like it process very easily.
Also the sulfite/nitrate salt quantity on the video i posted are for the anhydrous sulfite salt, the user that made the video also made another video
on how he made the sulfite salt and i think he is using the dihydrate like i did so the ratio is wrong. Probably why he has low yield(except from the
fact he has lost a lot of product from overheating the mix and a broken beaker/kept the top part).
I think this method is very promising.
For 5G of NaNO3 its 9.187G of CaSO3.
Na2S2O5+H2O→NaHSO3 NaHSO3+NaOH→Na2SO3+H2O Na2SO3+CaCl2→CaSO3+NaCl CaSO3+NaNO3→CaSO4+NaNO2 last one use heat - temp 230-300 C for 30-10 min.
[Edited on 14-8-2023 by fx-991ex]
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 198
Registered: 12-7-2018
Member Is Offline
|
|
I ve never tried this way but i think its for bigger scale lab. Production the best methode. You dont need to melt something and you can make it in
big Backers.
50 Parts Sodiumnitrate desolved in 150 Parts of Water. Add 350ml Ammonia solution with specific wight 0,96. After this give slowly 60 parts Zinc
powder to it and hold temperature between 20C and 25C.After an short time all nitrate is converted to nitrite.
Its from an patent who improved an old lab methode from Poggendorfs Analen of Physic and Chemistrie.
Concentrations and Temperatures are improtand if it isnt exactly followed the nitrate gets reduced to hydroxide.
|
|
|
Fantasma4500
International Hazard
Posts: 1682
Registered: 12-12-2012
Location: Dysrope (aka europe)
Member Is Offline
Mood: dangerously practical
|
|
Aluminium nitrite
aluminium metal doesnt react with nitric acid, and neither nitric acid vapors, but it will react with nitrate salts such as copper nitrate and iron
nitrate- however if aluminium can be kept seperate from a nitric acid / nitrate salt mixture the NO2 may react rapidly with the aluminium to form
aluminium nitrite
ideally the container is first flushed with CO2, butane maybe- as that is a very heavy gas
i did one attempt but the nitric acid/NOx fumes ate through the perforated plastic bag that was holding the shredded aluminium foil
(al foil flitter made by coffee grinding aluminium foil balls)
NO2 source was nitric acid and steel wool balls
i had a slight success proven by wettening the resulting mixture in IPAlcohol and adding dilute HCl which then formed IPNitrite, flame test faintly
showed IPNitrite but it was largely yellow, probably due to hydrogen effervescence and presence of sodium nitrate as i used Na2CO3 + HNO3 to form the
CO2 before i started the reaction, i shall attempt this further as this was a very poor attempt.
sadly it doesnt appear that aluminium carbonate exists, it decomposes into aluminium hydroxide which is difficult to filter as its a gel rather,
IPNitrite might be best way to scavenge the nitrite from this reaction
|
|
|
unionised
International Hazard
Posts: 5135
Registered: 1-11-2003
Location: UK
Member Is Offline
Mood: No Mood
|
|
Am I reading this correctly? The yield is about 50 micro moles per square cm per week.
So, with a 10cm by 10 cm electrode you would get less than a quarter of a gram of nitrite per week.
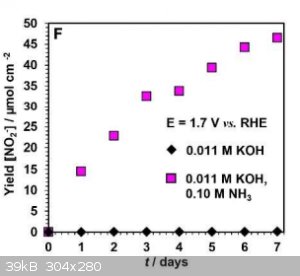
|
|
|
Kloberth
Harmless
Posts: 38
Registered: 2-6-2023
Member Is Offline
|
|
| Quote: Originally posted by Alkoholvergiftung | I ve never tried this way but i think its for bigger scale lab. Production the best methode. You dont need to melt something and you can make it in
big Backers.
50 Parts Sodiumnitrate desolved in 150 Parts of Water. Add 350ml Ammonia solution with specific wight 0,96. After this give slowly 60 parts Zinc
powder to it and hold temperature between 20C and 25C.After an short time all nitrate is converted to nitrite.
Its from an patent who improved an old lab methode from Poggendorfs Analen of Physic and Chemistrie.
Concentrations and Temperatures are improtand if it isnt exactly followed the nitrate gets reduced to hydroxide. |
Do you have a link to this patent or pdf?
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 198
Registered: 12-7-2018
Member Is Offline
|
|
See Attachment. They are in German.
Attachment: NitritDE000000168450A_2.pdf (65kB)
This file has been downloaded 370 times
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 198
Registered: 12-7-2018
Member Is Offline
|
|
First Part.
Attachment: NitritDE000000168450A_1 (1).pdf (78kB)
This file has been downloaded 380 times
|
|
|
Lionel Spanner
Hazard to Others
Posts: 175
Registered: 14-12-2021
Location: near Barnsley, UK
Member Is Offline
|
|
| Quote: Originally posted by unionised |
Am I reading this correctly? The yield is about 50 micro moles per square cm per week.
So, with a 10cm by 10 cm electrode you would get less than a quarter of a gram of nitrite per week.
|
That's with 0.1 molar ammonia, i.e. 0.17% w/v. I suspect a more concentrated solution would result in better yields.
[Edited on 22-8-2023 by Lionel Spanner]
|
|
|
Alkoholvergiftung
Hazard to Others
Posts: 198
Registered: 12-7-2018
Member Is Offline
|
|
I ve found this crusicible methode.Englisch and later german copy.
Very finely divided metallic copper is first prepared by distillation of acetate of copper, and from this freshly prepared metallic powder, 2
equivalents, or even a slight excess, are taken to 1 equivalent of nitrate, according to the equation
2 Cu + KO, NO⁵ = 2 CuO + KO, NO³.
Persoz took 320 grm. saltpetre and 200 grm. from the copper represented in the manner indicated.
To prepare an intimate mixture, the saltpeter is first dissolved in the smallest possible quantity of hot water, and then the copper is added, which
at first is difficult to wet. When the mixture has become quite uniform, it is heated in a porcelain dish, or better yet, in a cast-iron pan, in a
sand bath, stirring constantly to prevent splashing. When the mass is completely dry, there comes a moment when, like a pyrophor, it catches fire and
glows; when the combustion is over, which lasts but a moment, the reaction has taken place; it is allowed to cool, the melt treated with water,
filtered, and the nitrate allowed to crystallize. If excess copper has been used, no nitrate is present, and crystallized nitrite is obtained at once,
which is then melted and kept in well-closed flasks, as the salt is very hygroscopic. Any undecomposed nitrate of potash present is separated out by
the first crystallization, since it is far less soluble than nitrate of potash. The copper oxide obtained as a residue during the operation can, after
proper washing, be used for organic analysis, or at least for mixing with the organic substance to be examined, since, although it is very finely
divided, it is nevertheless very dense and to a much lesser degree hygroscopic than the annealing of nitrate of copper.
It is to be remarked, however, that common copper, even in a very finely divided form, would not be fit for the foregoing purpose, as in its
application
to bring about the intended reaction the temperature would have to be raised much higher, so that caustic potash rather than nitrate of potash would
be obtained; on the other hand, when using the copper prepared from the acetate salt, the reaction occurs at as little as 200° to 250° C. (Annales
du Conservatoire des arts et métiers, t. II p. 353.)
Man bereitet sich zunächst durch Destillation von essigsaurem Kupferoxyd sehr fein zertheiltes metallisches Kupfer, und nimmt von diesem frisch
bereiteten Metallpulver 2 Aequivalente oder selbst einen geringen Ueberschuß, auf 1 Aequivalent Salpeter, entsprechend der Gleichung
2 Cu + KO, NO⁵ = 2 CuO + KO, NO³.
Persoz nahm 320 Grm. Salpeter und 200 Grm. von dem auf die angegebene Weise dargestellten Kupfer.
Zur Herstellung eines innigen Gemenges löst man den Salpeter zunächst in der möglich geringsten Menge heißen Wassers, und setzt dann das Kupfer
hinzu, welches sich anfänglich nur schwierig benetzen läßt. Ist das Gemenge recht gleichartig geworden, so erhitzt man dasselbe in einer
Porzellanschale oder besser, in einer gußeisernen Pfanne im Sandbade unter beständigem Umrühren, um Spritzen zu verhüten. Ist die Masse
vollständig getrocknet, so tritt ein Moment ein, wo sie, gleich einem Pyrophor, Feuer fängt und erglüht; ist die Verbrennung vorüber, was nur
einen Augenblick dauert, so hat die Reaction stattgefunden; man läßt erkalten, behandelt die Schmelze mit Wasser, filtrirt und läßt das
salpetrigsaure Salz krystallisiren. Hat man überschüssiges Kupfer angewendet, so ist kein Nitrat vorhanden und man erhält sogleich krystallisirtes
Nitrit, welches man dann schmilzt und in gut verschlossenen Flaschen aufbewahrt, da das Salz sehr hygroskopisch ist. Etwa vorhandenes nicht zersetztes
salpetersaures Kali wird durch die erste Krystallisation abgeschieden, da es weit weniger löslich ist als das salpetrigsaure Kali. Das bei der
Operation als Rückstand erhaltene Kupferoxyd kann nach gehörigem Auswaschen zur organischen Analyse, wenigstens zum Vermengen mit der zu
untersuchenden organischen Substanz angewendet werden, da es, obgleich sehr fein zertheilt, dennoch sehr dicht und in weit geringerem Grade
hygroskopisch ist, als das durch Glühen von salpetersaurem Kupferoxyd erhaltene.
Zu bemerken ist indessen, daß gewöhnliches Kupfer, selbst in sehr fein zertheilter Form, zu dem vorstehenden Zwecke sich nicht eignen würde, da bei
seiner Anwendung
zur Hervorrufung der beabsichtigten Reaction die Temperatur weit höher gesteigert werden müßte, so daß man eher Aetzkali als salpetrigsaures Kali
erhalten würde; bei Anwendung des aus dem essigsauren Salze dargestellten Kupfers hingegen tritt die Reaction schon bei 200 bis 250° C. ein.
(Annales du Conservatoire des arts et métiers, t. II p. 353.)
|
|
|
woelen
Super Administrator
Posts: 8080
Registered: 20-8-2005
Location: Netherlands
Member Is Offline
Mood: interested
|
|
Interesting quote. The english translation contains some small, but essential errors. Below follows the corrected translation.
Very finely divided metallic copper is first prepared by distillation of acetate of copper, and from this freshly prepared metallic powder, 2
equivalents, or even a slight excess, are taken to 1 equivalent of saltpeter, according to the equation
2 Cu + KO, NO⁵ = 2 CuO + KO, NO³.
Persoz took 320 grm. saltpeter and 200 grm. from the copper represented in the manner indicated.
To prepare an intimate mixture, the saltpeter is first dissolved in the smallest possible quantity of hot water, and then the copper is added, which
at first is difficult to wet. When the mixture has become quite uniform, it is heated in a porcelain dish, or better yet, in a cast-iron pan, in a
sand bath, stirring constantly to prevent splashing. When the mass is completely dry, there comes a moment when, like a pyrophor, it catches fire and
glows; when the combustion is over, which lasts but a moment, the reaction has taken place; it is allowed to cool, the melt treated with water,
filtered, and the nitrite allowed to crystallize. If excess copper has been used, no nitrate is present, and
crystallized nitrite is obtained at once, which is then melted and kept in well-closed flasks, as the salt is very hygroscopic. Any undecomposed
nitrate of potash present is separated out by the first crystallization, since it is far less soluble than nitrite of
potash. The copper oxide obtained as a residue during the operation can, after proper washing, be used for organic analysis, or at least for mixing
with the organic substance to be examined, since, although it is very finely divided, it is nevertheless very dense and to a much lesser degree
hygroscopic than the annealing of nitrate of copper.
It is to be remarked, however, that common copper, even in a very finely divided form, would not be fit for the foregoing purpose, as in its
application to bring about the intended reaction the temperature would have to be raised much higher, so that caustic potash rather than nitrite of potash would be obtained; on the other hand, when using the copper prepared from the acetate salt, the reaction
occurs at as little as 200° to 250° C. (Annales du Conservatoire des arts et métiers, t. II p. 353.)
This is quite an interesting thing. I did not know that copper acetate can be converted to copper metal by heating the salt. This is definitely
something to try on a small scale. Maybe this process also works with NaNO3 instead of KNO3. The resulting NaNO2 is much less hygroscopic than KNO2.
Also interesting to see that in this old writing potassium ion is considered divalent. They wrote KO,NO5 for potassium nitrate, which we now would
write as K(NO3)2. Potassium nitrite is KO,NO3, which in modern notation would be K(NO2)2. Copper(II) at that time already was regarded as a divalent
ion.
|
|
|
| Pages:
1
..
13
14
15
16
17 |









