| Pages:
1
..
9
10
11
12
13
..
15 |
DougTheMapper
Hazard to Others

Posts: 145
Registered: 20-7-2008
Location: Michigan, USA
Member Is Offline
Mood: Energetic
|
|
Suppose the MEK was repeatedly dripped through a column of sodium percarbonate, all at less than 0C, thereby converting it to MEKP?
Does the H2O2 need to be aqueous to facilitate the reaction?
If that worked, one wouldn't have to worry about lots of acid loss. I don't know if the reaction would even take place without the acid catalyst
however.
|
|
|
497
National Hazard
Posts: 778
Registered: 6-10-2007
Member Is Offline
Mood: HSbF6
|
|
As far as I know the reaction requires acidic conditions to proceed. Then again I've never heard of anyone trying it in alkaline conditions...
I don't know if the reaction requires aqueous conditions, I've never seen it done otherwise.
I did have an idea that might reduce the amount of acid need: make a concentrated percorbonate solution and then cool it to 0*C or below, that will
reduce sodium carbonates solubility to below 7g/100ml, but I don't know if it would precipitate regular sodium carbonate hydrate or reprecipitate the
percarbonate. I imagine there is an equilibrium between precipitating hydrate or percarbonate, so it would depend on the concentration.. It is
manufactured by spraying anhydrous sodium carbonate with >55% hydrogen peroxide solutions.
|
|
|
Leander
Harmless
Posts: 28
Registered: 23-2-2008
Member Is Offline
Mood: No Mood
|
|
A friend of mine once concentrated some hydrogen peroxide to a concentration of ~55% and prepared TCAP with conc. H2SO4 as a catalist. The yield was
somewhat lower than the 'traditional' method with the exact same molar ratio's of H2O2/Acetone/Acid, only with a much higher water content. So my
guess is that the aqueous environment is necessary, although I can't really explain it scientificly.
[Edited on 24-8-2008 by Leander]
|
|
|
497
National Hazard
Posts: 778
Registered: 6-10-2007
Member Is Offline
Mood: HSbF6
|
|
In the conditions you just gave there would still be plenty of water present. Maybe less than normal, but still plenty, and things that require
aqueous conditions don't always require a lot of water, just a little.
I suppose if nothing happened when you mixed dry percarbonate with MEK, you could always try dissolving some water in the MEK...
I'm pretty sure the acid is required to catalyze the reaction though. I calculated a (room temp) saturated solution of percarbonate could make a 5%
H2O2 solution. You could try using NaHSO4 to acidify, it is cheap and OTC in large quantities. The resulting Na2SO4 has a low solubility at 0*C so
most of it could be removed by filtration before adding the MEK. That might not be a bad way to prepare H2O2 solutions in general. Through repeated
"dissolve percarbonate-acidify-filter-repeat" it may be possible to produce fairly concentrated H2O2. You may run into problem with impurities in
consumer percarbonate. Also some of it may be treated with 150*C air to decompose the H2O2 and create pressurized microbubbles of O2 in the crystals
that result in a desirable foaming action when dissolved.
There are MANY other perhydrates (aka peroxyhydrates) besides sodium carbonate perhydrate. See US patent 3140149, it has a pretty long list of them
(some new ones discovered since then). Even one salt, Na3PO4 for example, can have a large number of different perhydrates or perhydrate/hydrate
adducts. Some of them my be explosive on their own. I think the highest H2O2 content one I found so far is K4P2O7*7H2O2.
[Edited on 24-8-2008 by 497]
|
|
|
DougTheMapper
Hazard to Others
Posts: 145
Registered: 20-7-2008
Location: Michigan, USA
Member Is Offline
Mood: Energetic
|
|
Hello again -
I've finally rekindled my interest in chemistry, and I have a few questions I'd like someone with a more qualified opinion than myself to answer.
I plan to use HCl in place of the H2SO4 in this synthesis. Because HCl only has one protonizable hydrogen as opposed to H2SO4, do I need twice as much
to achieve the same acidity? (I've already compensated for the difference in concentrations.)
Also, it is extremely economical and convenient for me to purchase 3% H2O2 at the local dollar store. Unfortunately, this makes the reaction vessels
rather large and watery. After the 24 hour chill period, do you think it is possible to use a syringe to extract the large amount of acidic water
under the MEKP prior to the acid neutralization step? This would save me a lot of money in bicarbonate.
Also, instead of leaving the prepared MEKP out to lose water, what about washing it over some anhydrous MgSO4? I'd rather not have any accidents...
Perhaps I should store it in a container with a bag of MgSO4(anh.) suspended above the liquid.
Thank you very much,
DTM
[Edited on 23-5-2009 by DougTheMapper]
|
|
|
Leander
Harmless
Posts: 28
Registered: 23-2-2008
Member Is Offline
Mood: No Mood
|
|
The PH scale is based on the molar ratio of H+ in solution. 1 mole of H2SO4 gives 3 moles of H+, while 1 mole of HCl gives 1 mole of H+.
3% H2O2 is probably not going to work, because it dilutes the reactants so bad that the reaction simply doesn't runs to completion.
Separation is fully based on the density of the spent acid/H202 mixture. My Mekp actually floated on top of the spent acid, and then sunk again after
draining with water. When using concentrated chemicals the MEKP can simply be decanted off, and then neutralised. Neutralising all of the acid is
indeed quite a waste.
Further making large amounts of MEKP is really dumb. The stuff is so sensitive that 2-3 drops soaked in tissue paper can be set of with a 200 gram
hammer from 3cm height. I never made it again after discovering that.
[Edited on 24-5-2009 by Leander]
|
|
|
497
National Hazard
Posts: 778
Registered: 6-10-2007
Member Is Offline
Mood: HSbF6
|
|
| Quote: | | The stuff is so sensitive that 2-3 drops soaked in tissue paper can be set of with a 200 gram hammer from 3cm height. |
I think most of the danger can be avoided by only using plastic containers and tools to manipulate it. It is far harder to get it to detonate from
plastic on plastic impact..
|
|
|
DougTheMapper
Hazard to Others
Posts: 145
Registered: 20-7-2008
Location: Michigan, USA
Member Is Offline
Mood: Energetic
|
|
As always, I am extremely cautious. At no point will any MEKP be contained in glass or metal, and containers used will be spotlessly clean and not
have snap lids.
|
|
|
DougTheMapper
Hazard to Others
Posts: 145
Registered: 20-7-2008
Location: Michigan, USA
Member Is Offline
Mood: Energetic
|
|
Synthesis Complete
Having failed to locate a source for >3% H2O2 solution, I tried the following with much success:
460ml 3% H2O2
31ml MEK
20ml 31.45% HCl
I started by measuring all the reactants and chilling the acid and MEK in the freezer. I then simmered the H2O2 for several hours (without boiling) in
a clean container until only 75ml remained.
I chilled the 75ml H2O2 in the freezer. I then combined the cold H2O2 and MEK in a 200ml flask, and slowly stirred in the acid. The mixture
immediately clouded and large globules of insoluble material rose to the surface. I placed said flask in the refrigerator at 2 degrees C. After
several hours, the mix had separated fully, leaving a milky bottom layer underneath a clear top layer. A sample of the top layer was drawn for
analysis, and detonated with an earsplitting crack when hit with a hammer on an anvil.
I plan to wait the full 24 hours to let any remaining reactants do their job. At this point, I will draw off as much as possible from the flask and
deacidify it in a NaHCO3 bath. It will then be transferred to a container placed in a bell jar with anhydrous MgSO4 for desiccation.
Yield is TBD, but I would estimate c. 20ml. Further bulletins as events warrant.
-DTM
|
|
|
DougTheMapper
Hazard to Others
Posts: 145
Registered: 20-7-2008
Location: Michigan, USA
Member Is Offline
Mood: Energetic
|
|
Isolation Complete
Hello everyone, it's me again, and it's time for a follow-up.
I learned some lessons during the isolation process which I'll share for the benefit of future chem enthusiasts traveling this road.
First off, I'm pretty sure I used too much HCl. After all, it's only the catalyst. This caused big problems during neutralization when my bath became
nearly half a liter in volume to fully neutralize 25ml of MEKP. I doubled the acid content in the instructions so I would have the same hydrogen ion
concentration (H2SO4 vs. HCl), and paid for it in the end. I will try my next batch with half the HCl and post results.
I would also recommend neutralizing in a clean florence or volumetric flask, then adding water to push the MEKP (which floats) into the neck for
easier extraction. I was left trying to suck a 3mm layer of MEKP off 500mL of bicarb. solution in a beaker, which was neither fun nor efficient. I
ended up using a plastic paddle to dam up the MEKP as I poured the solution out from under it. I lost a bit that way, and MEKP has a strange habit of
sticking to glass. I lost a good amount on the walls of the beaker, not to mention the stench of all that exposed MEKP-covered surface area.
If done in the flask, I wouldn't expect odor to be problematic. In my unfortunate case, however, it was quite prolific. It smells like MEK mixed with
sage and pepper. It's quite unique.
Anyway, I managed to recover about 10mL after my smelly fiasco was over. I am certain, however, that I could have recovered at least 20 if not for
poor equipment choice.
One last note: everything with MEKP on it has to be rinsed in MEK (or some other solvent) to remove it. Water is completely ineffective. I have not
tried soap, but I plan to next synthesis.
The final product is as expected - detonates loudly when struck with a hammer, undergoes rapid deflagration when heated and unconfined in small
amounts. It resembles light mineral oil in both color and viscosity.
-DTM
|
|
|
grndpndr
National Hazard
Posts: 508
Registered: 9-7-2006
Member Is Offline
Mood: No Mood
|
|
Ill post my 2cents as having success with 3% Hydrogen peroxide.Yield would be considered atrocious but as used to get an first hand Idea as to its
properties It was a successful inexpensive lab.
That gave rise to other experiments,foremost an oxidizer, AN 100gr and MEKP 11gr/ml (OB) that could be a useful simple booster for difficult safety
exlosives.A simple rehash for sure but someone may percieve different uses, materials- addittions.
[Edited on 18-8-2009 by grndpndr]
|
|
|
User
Hazard to Others
Posts: 339
Registered: 7-11-2008
Location: Earth
Member Is Offline
Mood: Passionate
|
|
Indeed quite remarkable that 3% is strong enough to drive the reaction.
Not that i like organic peroxides but still I wonder if other explosive peroxides could be made using such low concentrations.
Also, often hydrogen peroxide in the stores arnt in a cool/dark place and could well be there for months , making the concentration even lower.
What a fine day for chemistry this is.
|
|
|
grndpndr
National Hazard
Posts: 508
Registered: 9-7-2006
Member Is Offline
Mood: No Mood
|
|
I would think a large turkey baster would work assuming the acid concentration didnt wreak havoc with the plastic components.
When down to the last of the liquor/mekp I placed it in a seperatory funnel.At the time 2000-2002 I was careless enough to stir not as carefully as I
should have with glass on glass containers /stir
stix.:Fortunately no Darwin award that year!My peroxide year
well say,little ap as Im a chicken at heart w/no heart for pressing AP into dets even though very littles required for mekp/AN.Last of the peroxides
for me but I did like the MEKP,the idea of a liquide explosive added to AN reminded one of a kinepack mix, sadly the brisance wasnt there.Neither is
the AN/NM easily available anymore
[Edited on 22-9-2009 by grndpndr]
|
|
|
DougTheMapper
Hazard to Others
Posts: 145
Registered: 20-7-2008
Location: Michigan, USA
Member Is Offline
Mood: Energetic
|
|
Second Synthesis
I use a 10mL syringe with a curved tip that I got when I had my wisdom teeth removed. I, as with grndpndr, am a chicken when it comes to sensitive
stuff like this. I work behind quite a shield.
Anyway, I isolated the stuff for the second time a few days ago, this time in a round-bottomed flask. The reaction bath was about half of this 250mL
flask, and the neutralization solution was added in excess until the MEKP was forced into the neck of the flask. extraction was a breeze, and yield
was considerably higher than my last iteration. Oh, and I also used HALF the HCl this time with no perceptible change in yield.
The flask is covered in condensation. The MEKP was foggy until it sat in the orange collection vial for about a day, where it became totally clear
with a small bead of water at the bottom, which was removed with the syringe.
I have recently acquired about 40g of AN, and I plan to make an 8:1 AN/MEKP charge this weekend. Pics and vids will be available.

[Edited on 9-10-2009 by DougTheMapper]
|
|
|
grndpndr
National Hazard
Posts: 508
Registered: 9-7-2006
Member Is Offline
Mood: No Mood
|
|
Theres a topic in reagents and appaatus regarding concentrating H2O2.from what Ive gathered unless your shooting for unnescessarily high % for these
peroxide synthesis the simplest method is likely freezing.It has been in my limited experience.It will likely involve some guesswork as to the
ultimate concentration at least in my case but I would think it would be relatively accurate/effective to 30-35% and higher,maybe not advisable for us
beginners(higher 35%H2O2)That at least would cut down on quantities
involved as well as improve yield guite a bit(over 3%H2O2) but still a very dangerous product the result.
And as the years have flown by I have become increasingly attached to my extremitys so I personally wouldnt try peroxide labs any longer although I
eagerly read others 'adventures' .Regards
[Edited on 19-11-2009 by grndpndr]
|
|
|
gnitseretni
Hazard to Others
Posts: 282
Registered: 5-1-2007
Location: Colombia
Member Is Offline
Mood: No Mood
|
|
At what concentration does H2O2 leave a stain on your skin? 35% H2O2 leaves a stain. Perhaps after you froze 3% H2O2 a couple of times, put a drop of
it on your finger to see if it'll leave a stain.. if so, use it and report your yeild; If not, freeze it until it does leave a stain.
Dunno - might work, no?
|
|
|
grndpndr
National Hazard
Posts: 508
Registered: 9-7-2006
Member Is Offline
Mood: No Mood
|
|
Id prefer to not use my skin as a sort of litmus paper.The 60% or thereabouts Ive heard possible by freezing probably wouldnt be to healthy for living
tissue.In the past Ive frozen a known quantity measured what remained and subtracted a bit for loss.Probably close enough though Im sure theres a
method to accurately measure %.
|
|
|
gnitseretni
Hazard to Others
Posts: 282
Registered: 5-1-2007
Location: Colombia
Member Is Offline
Mood: No Mood
|
|
Oh well, just a thought 
|
|
|
Microtek
National Hazard
Posts: 869
Registered: 23-9-2002
Member Is Offline
Mood: No Mood
|
|
Just do a redox titration of the concentrated peroxide using a suitable indicator.
One possibility would be to use a solution of KMnO4 (of known concentration) to oxidize the peroxide. If the reaction is acidified with sulfuric acid,
the permanganate ion goes quantitatively to Mn(II) which is colourless. So the endpoint is indicated by the persistence of the purple colour.
If you want decent precision, you will have to prepare the KMnO4 soln carefully, as there is always some MnO2 in the KMnO4 you buy (no matter the
grade). If you intend to do the titration immediately, you can just standardize the soln on sodium oxalate with reasonable results, but you won't be
able to use the soln the next day.
|
|
|
Zinc
Hazard to Others
Posts: 472
Registered: 10-5-2006
Member Is Offline
Mood: No Mood
|
|
Sorry to bring up an old thread, but I belive this is the best one for the question I have.
Hww stable is the 50:50 MEKP/AP mix in mildly basic conditions?
I ask because I have some AN that is slightly basic (I belive due to MgO contamination), so I am wondering how safe is it to mix it with the MEKP/AP.
I know that in the short run (an hour or two of contact nothing happens, at least nothing observable) But what about 24-48 hours?
|
|
|
Bot0nist
International Hazard
Posts: 1559
Registered: 15-2-2011
Location: Right behind you.
Member Is Offline
Mood: Streching my cotyledons.
|
|
Has anyone experimented with MEKP on C<sub>6</sub>H<sub>7</sub>(NO<sub>2</sub><sub>3</sub>O<sub>5</sub>. I did a search <a
href="http://www.sciencemadness.org/talk/search.php?token=&srchtxt=mekp+nitrocellulose&srchfield=body&srchuname=&f[]=all&srchfrom=
0&filter_distinct=yes&searchsubmit=Search">here</a> and <a
href="http://www.sciencemadness.org/talk/search.php?token=&srchtxt=gun+cotton+MEKP&srchfield=body&srchuname=&f[]=all&srchfrom=0&am
p;filter_distinct=yes&searchsubmit=Search">here</a> but found nothing.
I tried it yesterday, but the MEKP was still a little "wet" with mek and it melted the cellulose nitrate. After letting the mekp dry a bit it seemed
to withstand it without going all gooey. I would love to experiment more, but I get very nervous when making mekp and restrict myself to about 3- 5 ml
in those little HDPE test tubes, so I haven't been able to run a good control series to compare to. I have 5ml drying now and will try to get around
to doing some comparisons tonight.
My mekp was made with 15% H<sub>2</sub>0<sub>2</sub>, 95% H<sub>2</sub>SO<sub>4</sub>, and pure mek
with the proportions adjusted for concentrations.
The cellulose nitrate was double nitrated in a H<sub>2</sub>S0<sub>4</sub> +
NH<sub>4</sub>NO<sub>3</sub> using 55ml to 31g and 5 grams of cellulose. 3 hour nitrations with agitation each time.
|
|
|
grndpndr
National Hazard
Posts: 508
Registered: 9-7-2006
Member Is Offline
Mood: No Mood
|
|
eagerly awaiting results of your experiments-would particularly like to hear of your perception or actual results on test plates etc.
Off topic, it was asked but not answered a few pages back
concerning a chlorate as an oxidizer for MEKP.Specifically sodium chlorate as the oxidizer was the question but Id like to hear if there were any
contraindications to using k chlorate,ammonium perchlorate etc.Theoretically of course.
|
|
|
trezza
Harmless
Posts: 23
Registered: 23-5-2009
Location: Australia
Member Is Offline
Mood: ok... I guess
|
|
I've experimented with MEKP before.
-------------------------------------------------------------------
Synthesis of Methyl Ethyl Ketone Peroxide using HCl
-------------------------------------------------------------------
Chemicals used:
38mL 30% Hydrogen Peroxide
25mL 100% Methyl Ethyl Ketone Peroxide (PVC Primer solution)
15mL 30% Hydrochloric acid
5-10% Sodium Hydrogen Carbonate solution
Everything was chilled in the freezer and an ice water bath was prepared before I started the reaction.
The H2O2 and MEK were mixed in a 250ml beaker and placed in the ice bath, then over a period of 10 minutes the HCl was added dropwise to the solution,
the temperature was watched closely and kept below 5 degrees C.
Once all of the acid was added I moved the beaker to the refrigerator still in the ice water bath for 18 hours.
When the beaker was removed there was an oily layer layer on the bottom and a thin oily layer on top of the solution.
200mL of Sodium bicarbonate solution was placed in a 1L beaker and the MEKP solution was poured into that, there was some frothing and fizzing, I
stirred it for 10 minutes until the MEKP layer had been broken up a few times and the fizzing stopped. I collected the MEKP in a syringe and left it
in a large beaker to dry.
Final Yield was about around 10mL.
---------------------------------------------------------------------
Testing: Methyl Ethyl Ketone Peroxide/Potassium Nitrate
---------------------------------------------------------------------
I have seen TATP/AN combinations used to detonate ANFO
I had also heard of potassium nitrate and TATP mixtures giving semi successful results so I decided to try MEKP/PN
I wanted to find a good ratio so I decided to Balance the oxygen, I used this equation.
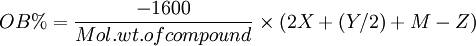
X represents the number of Carbon atoms, Y represents the number of Hydrogen atoms, Z represents the number of Oxygen atoms. M represents the number
of metal oxide molecules which can be made from 1 molecule of each compound.
Here I made a graph which shows the balance of oxygen for several energetic compounds...

From that I can tell that a perfect ratio of ammonium nitrate to nitromethane (NM) would be 66:33 which sounds correct as people seem to use mixtures
ranging between 25:75 and 50:50
My working out is attached in a .txt file below.
Now to business. I used a ratio of 1mL:4.5g of MEKP to Potassium Nitrate which gave a consistency similar to damp sand, this was placed in a cardboard
tube and 1g of HMTD was used to detonate it. There was quite a loud bang, it was more like a thud, similar to what I would expect from an ANFO
detonation, it certainly seemed more powerful than 1g of HMTD on its own so I could gather the MEKP detonated. Afterwards I checked for any sign of
the Potassium nitrate on the ground but I couldn't find anything, I'm fairly sure the MEKP/PN detonated completely.
[Edited on 8-3-2011 by trezza]
Attachment: oxygen balances.txt (2kB)
This file has been downloaded 1008 times
|
|
|
Bot0nist
International Hazard
Posts: 1559
Registered: 15-2-2011
Location: Right behind you.
Member Is Offline
Mood: Streching my cotyledons.
|
|
I will have those test results as soon as I can figure out a good way to quantify the deflagrations, as I would hate to just present you all with just
my perceptions.
My first thought was to use a poster that had one inch black increments in a + pattern. I mounted the cotton on a wire so that it was suspended in the
center of the plus. I then thought I could use my camera to determine the size of the resulting fireball, like on mythbusters. That failed because my
camera cant handle the extreme light and just goes all white at the moment of initiation.
My next idea was to place the 'charges' on a piece of foil that rests on top of my digital scale. I planed on taring it out, and then measuring the
sudden increase in pressure on the scale. This also failed, as the spikes happened to fast for the scale to accurately measure them.
I can't really think of a good way to quantify the deflagrations using the tools I have, so I will re-due the tests and post some pictures/short
videos of the deflagrations, along with my personal observations. That isn't the most scientific approach, but I am at a loss.
I'll synthesis a few more millimeters tonight and I will try to have some meaningful results up by the weekend.
|
|
|
Chordate
Hazard to Others
Posts: 108
Registered: 23-2-2011
Member Is Offline
Mood: No Mood
|
|
Back in the 1500s, when star forts were in vogue, a lot of effort went into zeroing cannons within fortifications, such that if one of these forts was assaulted the
artilleryman would know at exactly what angle to align their cannons in order to hit an attacking force at important ranges relative to the
surrounding fortifications.
The problem then became variable gun-powder quality, the way they tested this was to place a cannonball of known weight in an iron cup next to a
metered pole, then ignite a measured amount of gunpowder underneath it and see how high the ball went. Its crude, but it would be do-able. Calculating
the amount of energy released accurately wouldn't be trivial, but if you had a reference sample of known qualities you could calibrate that scale and
then work from there.
|
|
|
| Pages:
1
..
9
10
11
12
13
..
15 |









