| Pages:
1
2
3
..
7 |
CycloKnight
Hazard to Others

Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Amino alcohol via Akabori, trial run
Recently I’ve been experimenting with the Akabori reaction, just curious to see if the process for making amino alcohols can be improved upon. I
thought it would be quite an interesting amateur science experiment.
So far its been an interesting challenge !
The Akabori refers to the reaction between an aldehyde & amino acid to yield the corresponding amino alcohol.
For this run, the following chemicals were used:
83 g l-alanine (considerable excess)
40g benzaldehyde
250ml xylene as solvent (convenient & OTC)
The conventional low yielding approach is to simply heat benzaldehyde & l-alanine at 150-160 C until CO2 evolution ceases.
The objective with this variation is two fold: to keep the l-alanine in excess at all times by slowly dripping the aldehyde into the reaction mix,
the idea being that this will increase yields wrt to consumed benzaldhyde, & to keep water out of the reaction.
When water gets into the reaction, by-products are immediately formed and the reaction mix will turn red (tar) quite quickly. The boiling temp of
xylene is 139 C, therefore if water drips back into the boiling solution, the results can send foam as far as the condenser & beyond.
Foaming is another issue with this particular trial run, higher dilution is required, higher than what was used in this trial run. By the end of the
reaction, due to tar formation, foaming was getting a bit out of control.
The reaction was maintained at high reflux to help flush out generated moisture, this was maintained for 1 hour before the aldehyde addition. To
assist with this task, a DIY Dean Stark trap was hastily thrown together, to collect generated water and help keep it out of the mix.
First, the l-alanine was finely powdered as much as possible.

Vegetable oil was used for the oil bath. Stir bar spinning.

General setup.



Moisture starting to collect in the trap.


For the first aldehyde addition, 26 ml of aldehyde was slowly dripped into the solution. Later (see below), an additional 14 ml was added; 40 ml
benzaldehyde added in total.



Aldehyde addition is begun, 1 drop every 2 seconds.

Byproducts collecting, the red drops are water.



1 hour after the addition. The addition of the 26 ml aldehyde took 1 hour 15 min to complete.

The reaction was run until water stopped condensing, then the addition of the rest (14 ml) of the aldehyde was begun.

Benzoic acid forming in the aldehyde dropping funnel…

2 hours after the aldehyde addition was started.



Total collected condensate at the end of the reaction

Water accumulation ceased, so the reaction was stopped.
Reaction time = 4 hours.
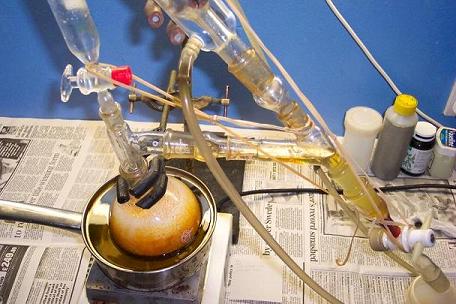

The reaction mix was put in a freezer to cool, and precipitate impurities.
Once no more impurities (l-alanine mostly) were seen to fall out of solution, the mixture was carefully removed, 3* 50 ml cold toluene was used to
rinse the unreacted l-alanine, the pooled toluene extracts were then shaken with 15% HCl until the acid remain acidic upon settling. Temperature
before acid addition was 10 C, after addition the temperature had increased to 26 C. A little extra dH2O was added to help with the interphase mixing.
Tar has settled to the bottom, aqueous layer is in the middle, with toluene/xylene on top.
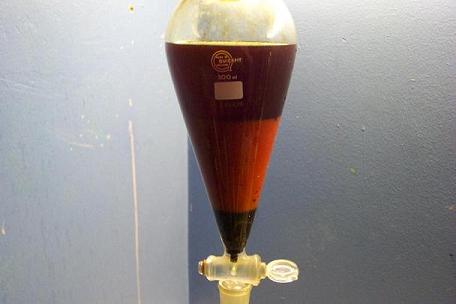
Tar dripped out.

Aqueous fraction extracted 5 times with 50 ml toluene. 10 minutes settling time between each extraction

Followed by 5 extractions with 50 ml dichloromethane

Followed by 5 extractions with 50 ml ethyl acetate.

Aqueous solution was then placed in a pyrex dish and placed on a small pot boiling water to maintain 100 C until the level of the solution had boiled
down into a paste. The paste was kneaded with a spatula until the HCl odour had mostly disappeared.


Ethyl acetate has been found to work well as solvent for crystallising the final product (Eureka, finally a means of cleaning up the goop.).
Ethyl acetate washing up. Don’t do as shown – its unnecessary and more difficult, just place the paste in a suitable container and then mix in the
ethyl acetate (approx 80 - 100 ml).

The ethyl acetate soaked paste is poured into a 250 ml RBF and heated until all dissolved, then set aside to slowly cool.

After 8 hours, no more crystals appear to be forming. The flask was then put in the freezer for a few more hours.
The crystal mass fills half the flask, not only lining the glass edge as shown in the picture.



Then as fate would have it, luck ran out. Although the acetone used for the washup had been dried with a couple hundred grams of anhydrous magnesium
sulphate, the crystals still dissolved somewhat. This was only realised after the acetone extracts were placed in the same container as the previous
run’s acetone extracts, so I can't work out the exact yield (Doh!).
It would appear that either: the PPA.HCl salt (phenylpropanolamine.HCl aka norephredine.HCl) is slightly soluble in acetone, or the acetone was wet.
SWIM has no idea, yet.
Either way, the dried yield was found to be 10 grams (not including the amount still in the acetone).
Judging by previous runs, the actual yield is probably at least several grams higher.
In the workup it was obvious that some benzaldehyde has remained unreacted, indicating that a higher dilution should be used in future runs.
Any suggestions on how to improve this process further?
Additional note: the ethyl acetate used was distilled from "acetone free" nail polish remover, and was later found to still contain a little isopropyl
alcohol.
[Edited on 10-6-2006 by CycloKnight]
|
|
|
Vitus_Verdegast
Hazard to Others
Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
By Jove!! Is that the Daily Mail you're reading ?! 
Just kidding.... Good work!
I'm afraid I haven't read up enough on this reaction to give any suggestions, only the reports of failure that were posted a couple of years ago by
somebody named 'DRIVEN'.
I think Beilstein should be able to give you PPA solubility info or at least where to find it.
[Edited on 10-6-2006 by Vitus_Verdegast]
Sic transit gloria mundi
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Akabori runs
........only the reports of failure that were posted a couple of years ago by somebody named 'DRIVEN'.
Its easy to carry out a sucessful reaction, the workup however is quite tricky.
The general procedure for amine extraction cannot be used here. The general procedure being:
-extraction of final reaction mix using dilute HCl
-basification with 25% NaOH solution
-extraction with organic solvent
-washing organic extacts to remove dissolved NaOH
-drying & gasification with HCl to give the amine salt
-washing the salt in suitable solvent
The reason is mainly because this freebase dissolves quite well in water, prohibiting the organic freebase extracts from being washed prior to
gasification, resulting in lots of NaCl formation (and other garbage) in the product. The dissolved NaOH also prohibits the use of vacuum distillation
for distillation of the freebase directly, since the NaOH will destroy it at the temperatures required for the vac distillation.
If the freebase/organic solvent mixture is washed to remove the NaOH, the product ends up in the aqueous washes and is very difficult to remove.
Even if the aqueous washing step is skipped in the general procedure, the final crystallised product (and salt!) forms a suspension in the gassed
toluene solution, and forms an orange soup-like mix that still requires further purification.
The coloured impurities remaining from that step do not dissolve well in toluene, but do in acetone (with some loss of product).
If one chooses to use the photo procedure posted earlier and skip the HCl gassing/freebase step, then the final brown paste cannot be purified using
acetone alone. Acetone was used only to purify the ethyl acetate soaked crystals (BTW, this was because SWIM had run out of dried ethyl acetate)
The use of ethyl acetate for final crystalisation has given the best results seen so far, definitely a step in the right direction for OTC workup.
The more conventional procedure is listed in Chemical Abstracts, Volume 47, column 3347 uses N-methyl-d,l-alanine instead for the synthesis of
d,l-ephedrine. In that procedure they use 50 grams of benzaldehyde & 20 grams of the amino acid, mixed together and heated (without solvent). 12 g
product are claimed.
When that procedure is used with alanine there is little or no benzaldehyde recoverable from the mix, it is simply mostly converted to tar, PPA,
water & CO2 during the reaction.
With the xylene method, about half of the benzaldedhye is recoverable from the solvent !!
Therefore, taking only the REACTED benzaldehyde into account, the reaction efficiency is increased somewhat.
On the other hand, recovering the aldehyde from the xylene mixture by vacuum distillation isn't exactly ideal so what SWIM is aiming to find is the
optimal dilution & reaction time that ensures that all the aldehyde is reacted thus giving (in theory at least) the highest reaction efficiency,
without the hassle of recycling unreacted aldehyde.
During the xylene run posted ealier, solution colour began to change from light brown to orange after ~20 ml aldehyde had been added. It also appeared
that the last 10 ml (30-40 ml addition) aldehyde was simply getting converted to tar.
This could indicate a target dilution, as well as an aldehyde to amino acid ratio, to aim for in order to decrease aldehyde consumption wrt yields.
As for product purity, this is what is known so far:
1) The light cream coloured product does react in warm NaOH solution to yield a light oily layer floating on top, posessing a very distinctive ammonia
freebase odour.
2) The melting point is quite high, but hasn't been measured yet. On a stainless steel spatula, it takes a candle flame about 8 seconds before any
melting is noted, which certainly doesnt rule out the mp being near the documented value of 195 C.
3) posesses a strong saline-bitter taste
4) if the freebase is acidified with H2SO4 to form the sulfate, and fed into 75% sulfuric acid at 145 C in the presence of ZnCl2 catalyst, the steam
distilled product contains the corresponding ketone. This was confirmed by the formation of the bisulfite addition product. This would appear to
confirm that the main product is indeed phenylpropanolamine.
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
I feel that identification of this as PPA is premature. TLC? MP of the freebase?
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Product composition.
The freebase hasn't yet been isolated on its own in sufficient purity for a mp test. Any suggestion(s) on how to do it?
How about an accurate mp test for the salt?
I'm not quite sure what else aside from PPA could produce the ketone according to this process:
http://designer-drugs.com/pte/12.162.180.114/dcd/chemistry/p...
[Edited on 11-6-2006 by CycloKnight]
|
|
|
Ramiel
Vicious like a ferret
Posts: 484
Registered: 19-8-2002
Location: Room at the Back, Australia
Member Is Offline
Mood: Semi-demented
|
|
I hate to be off topic CycloKnight (it looks like there's a good chemist here), but that's 37 pictures there... omg bandwidth! Someone pays for that
megabyte or so for each person who view this page. In this case, perhaps a picture is worth omitting for a few concise descriptions, eh?
Caveat Orator
|
|
|
Polverone
Now celebrating 21 years of madness
Posts: 3186
Registered: 19-5-2002
Location: The Sunny Pacific Northwest
Member Is Offline
Mood: Waiting for spring
|
|
Actually, we're fine as long as we don't go over the 240 GB or thereabouts that we're allowed each month. Below the threshold, it's a fixed rate
regardless of bandwidth used.
PGP Key and corresponding e-mail address
|
|
|
Vitus_Verdegast
Hazard to Others
Posts: 297
Registered: 5-12-2004
Location: Ottoman Empire
Member Is Offline
Mood: tea time
|
|
My Beilstein access is gone for the moment, does anyone have any information whether this reaction can be performed succesfully on substituted
benzaldehydes?
(I recon there should be no problems)
Sic transit gloria mundi
|
|
|
S.C. Wack
bibliomaster
Posts: 2419
Registered: 7-5-2004
Location: Cornworld, Central USA
Member Is Offline
Mood: Enhanced
|
|
It has been done with piperonal and N-methyl alanine, no yield given in CA. IIRC all three of the CA abstracts that I am aware of on this (this is the
only one with a substituted benzaldehyde and (N-methyl) alanine) are translations from Japanese, a patent and two J. Pharm. Soc. Japan articles.
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Update
The yield of PPA given in the previous reaction was no more than about 50% of the final crystallised product.
So far a few different variations on this reaction have been attempted.
As solvent, white spirits works quite well.
Reflux was maintained at 165 C while the aldehyde was slowly added. But the reaction time was still quite high, 4 hrs minimum.
Overall, its been found that the lower temperature variations appear to be producing a larger proportion of other amines, most notably benzyl amine.
The best approach to use for this reaction, is the conventional one (without solvent).
This yields a higher proportion of the desired amine, and is far less time consuming.
Until someone finds a way to minimise the unwanted amine by-products, this is how its done.
Conventional akabori run.
200 ml benzaldehyde
100g l-alanine
equipment required: distillation apparatus & oil bath, hotplate w/stirrer
Simply set up the equipment for distillation, generated by-products will distill out of the reaction as they form.
Finely ground alanine is first added, then the aldehyde.
It's essential to keep the stir bar spinning for the duration of this reaction.
Oil bath temperature is heated to 150-160 C and maintained at this temperature until the evolution of CO2 diminishes (>1.5 hrs).




Reaction time ~ 2 hrs.
Extract the final reaction mix with an equal volume of toluene.
Wash the unreacted alanine a few times with a small volume of toluene.
Combine the toluene extracts and set aside to cool, separate the toluene from any precipitate that settles.
Wash the combined toluene extracts with dilute HCl to remove the amine(s).
Wash the aqueous amine solution with DCM to clean it up as much as possible. This is quite tedious and prone to emulsion forming, especially with
toluene.

Basify by slowly adding NaOH granules.

Top layer is mostly freebase ~80 ml.
The freebase is extracted with organic solvent and carefully washed with several ml of distilled water to remove most of the NaOH.
Solvent is evaporated. Residue double distilled under vacuum to yield 35 grams PPA freebase. Melting point ~50 C.
If a way can be found to efficiently extract the remaining PPA from the other distillation fractions, then any remaining PPA could be recovered.
Overall, this was far less work than the previous runs using solvent. Not too impractical for producing a little PPA if you happen have benzaldehyde
lying around.
[Edited on 23-6-2006 by CycloKnight]
|
|
|
mr_orange
Harmless
Posts: 2
Registered: 24-6-2006
Member Is Offline
Mood: No Mood
|
|
Wow, read science madness off and on, but this thread floored me.
A thousand thanks for sharing the results and pictures of your experiments.
P.S.---Some of the guys over at www.synthetikal.com have been discussing this, it'd be nice if you joined us.
\"It\'s like baking a cake, where one of the ingredients is a hand grenade\"
|
|
|
Devilfish
Harmless
Posts: 3
Registered: 5-7-2006
Member Is Offline
Mood: No Mood
|
|
SWIM tried this way as well some months ago but adding NaOH he shot the pH too high, not knowing that this will destroy the PPA. Yielded some beakers
clotted with a really sticky mass. It took hours to get the labsware clean again...
but reading this writeup SWIM had the idea, that it could worth a try to substitute the workup with a steamdestillation of the FB...
[Edited on 5-7-2006 by daFlo]
|
|
|
Maja
Hazard to Others
Posts: 143
Registered: 27-2-2006
Member Is Offline
Mood: No Mood
|
|
From one forum, one guy told that benzaldehyde can be replaced with other aldehyde as piperonal to get MDPPA and then to treat it same way as PPA
freebase to get P2P,but in this case we would get MDP2P. Maybe you have tried it and/or have refs / patents ? I WOULD be very grateful
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
| Quote: | Originally posted by Maja
From one forum, one guy told that benzaldehyde can be replaced with other aldehyde as piperonal to get MDPPA and then to treat it same way as PPA
freebase to get P2P,but in this case we would get MDP2P. Maybe you have tried it and/or have refs / patents ? I WOULD be very grateful
|
No refs, have not tried the MD variation - but it sounds interesting. I'd imagine the yields would be quite low.
I wonder if the piperonyl could be fed to the Neff reaction with nitroethane to give the substituted PPA?
Can say that the P2P approach is much simpler, much easier workup. Basically with the P2P variation, the amine mixture is converted to the sulfate
with a little sulfuric acid, and the whole mix fed to the high temp acid (with ZnCl2 catalyst or other catalyst) and the ketone steam distilled.
Droplets of ketone began distilling almost immediately. Quite easy, really.
The PPA reacts, and all the other garbage just gets left in the pot.
What doesn't steam distill (other amines left unconverted) is simply discarded. From 360 ml alehyde & equivalent amount of l-alanine, yielded
about 65 ml free amine(s). On the first acid hydrolysis run SWIM attempted, this then gave at least 35 ml of ketone AFTER being vac distilled.
Basically, the whole mix was added at once, higher yields would probably be gained if an amine.sulfate/sulf. acid mixture was dripped in during the
steam distillation.
That yield of ketone is higher than the amount of PPA that SWIM has ever managed to reclaim from a reaction of that scale.
Trying to isolate pure PPA from the mixture of amines is quite tedious and doesn't appear too efficient either. There may be azeotropes forming with
the PPA too, still haven't been able to work that one out. Also, if there is are significant oily impurities in the PPA, it will not crystallise at
room temperature and will require redistilling.
| Quote: | Originally posted by DaFlo
SWIM tried this way as well some months ago but adding NaOH he shot the pH too high, not knowing that this will destroy the PPA. Yielded some beakers
clotted with a really sticky mass. It took hours to get the labsware clean again...
but reading this writeup SWIM had the idea, that it could worth a try to substitute the workup with a steamdestillation of the FB...
|
Definitely be careful with the NaOH, SWIM was quite apprehensive about it at first. Maybe try using sodium carbonate instead of NaOH. Either way, the
NaOH needs to be washed out at much as possible prior to the vac distillation to prevent the product degrading to tar at the high temperatures. Some
will inevitably remain and destroy some of the product before it can vac distill. Never managed to do a run without at least a teaspoon full of dark
red tar remaining, SWIM always wondered what that tar might have been if a stronger vacuum had been used...
The only problem I can see with the steam distillation variation is with reclaiming the product from the water. Maybe it would work if the the PPA
laden water were acidified to form the salt and then the water boiled off using a short packed column to minimise losses?
|
|
|
Punk
Hazard to Self
Posts: 64
Registered: 22-2-2006
Member Is Offline
Mood: No Mood
|
|
Looks to be a lot of work how does the yeild compare to simply heating the 2?
|
|
|
Devilfish
Harmless
Posts: 3
Registered: 5-7-2006
Member Is Offline
Mood: No Mood
|
|
why using a column, when boiling off the water? the PPA is HCl-salt at this moment and won´t form an azeotrope with the water. only the freebase
should do so or am I wrong?
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
| Quote: | Originally posted by daFlo
why using a column, when boiling off the water? the PPA is HCl-salt at this moment and won´t form an azeotrope with the water. only the freebase
should do so or am I wrong? |
No azeotrope with the salt. You''ll have say 35 grams of product in a relatively lare quantity of water. As you boil it down, your product
concentrates and much of your product will carry over with the steam.
|
|
|
Devilfish
Harmless
Posts: 3
Registered: 5-7-2006
Member Is Offline
Mood: No Mood
|
|
erm... having a solution of ephedrine.HCl or meth.HCl in water and boiling the water away won´t lower the ammount of ephedrin/meth.HCl remaining.
Why should PPA do so???
|
|
|
CycloKnight
Hazard to Others
Posts: 128
Registered: 4-8-2003
Member Is Offline
Mood: Still waiting for the emulsion to settle.
|
|
Sorry maybe I should have explained that better.
I was only referring to aerosol carry over. Using a short column (prefereably with packing) helps eliminate the carry over, thats all.
And yes ephedrine HCl and meth HCl will also carry over.
Losses aren't that high, but if youre boiling down a dilute solution of your product, its easy to lose 20% or more of your product.
I.e. if you were to take 5 grams of table salt and dissolve it in a gallon of water, then boil it down to dryness without a short column (or similar),
you'd be very lucky if you were able to recover 4 grams. The yield would decrease with increased boiling rate.
[Edited on 11-8-2006 by CycloKnight]
|
|
|
Punk
Hazard to Self
Posts: 64
Registered: 22-2-2006
Member Is Offline
Mood: No Mood
|
|
added trials
L-alanine/benz-->norpseudoephedrine freebase
Basicly the 2 were heated directly, diluted with a huge access of water, filtered out any solids, made basic with sodium bicarbinate and chilled. An
expected amount of white fluffy crystals settled to the bottom whare they were filtered and washed with cold water.
next step hydrolisis
Anyone have suggestions on obtaining the highest yeild from a norpseudoephedrine--> norephedrine via hydrochloric acid hydrolisis ?
Ive read that only 50% of the starting material can be tranformed. Could someone explain why?
Why is there a strong sulfer smell when boiling norpseudoephedrine hcl in hydrochloric acid?
Could certain molar ratios be used to control the problem of the norephedrine shifting back to norpseudoephedrine?
Is there a feasable way to seporate norpseudoephedrine from norephedrine?
[Edited on 15-8-2006 by Punk]
|
|
|
IOC
Harmless
Posts: 5
Registered: 29-7-2006
Member Is Offline
Mood: No Mood
|
|
Try alanine/benz, heat directly, when CO2 is done add equal amount of toulene then add equal amount of H2O and acidify to PH 4, seperate water phase
and 3 x methylene chloride to clean.
Boil down H2O pahse to a brown paste then add dry acetone to leave clean white powder.
Havnt had time to qualify MP, next time though
IOC......out
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
And what exactly makes you believe that without removing alanine your crude product is not mostly - guess what? – an alanine salt!
You need to wash with NaOH or Na2CO3 solution to remove the left over alanine before you extract any potentially formed 1-phenyl-2-aminopropanol.
Using only water to wash off the alanine would reduce the yield since alanine can act as an acid and will take considerable amounts of
1-phenyl-2-aminopropanol with it into the washes (1-phenyl-2-aminopropanol is even by itself slightly soluble in water, but when protonated is taken
into the water phase completely).
This might also explains why CycloKnight got such low yields in the first attempt (I mean the one described in the first post). He should have made
the postreaction mixture basic. Then he would not have to extract the water phase 5 times and still get so little product (assuming the conversion was
better than what it looks like).
The work up in his latest solventless attempt is more exemplary, though it would be worth to try another approach as well. Some of that alanine
precipitated from toluene might have well contained some 1-phenyl-2-aminopropanol absorbed as its toluene insoluble salt with alanine. If instead of
removing the precipitate, it would be dissolved by adding enough 20% NaOH to the toluene diluted postreaction mixture, the yields of the
1-phenyl-2-aminopropanol extracted in the toluene might be possibly improved and absolutely all alanine removed (of course, if this issue really is a
major problem).
However, I do think you are all underestimating the acidic properties of amino acids. It is true that their solutions are only negligibly acidic due
to their ambiphilicity, but they can and will behave as acids toward bases of strength comparative or grater to that of the alpha-carboxylic
amine group. 1-phenyl-2-aminopropanol is only very slightly less basic than alanine (pKa's of conjugated acids: 9.44 [PPA] vs. 9.69 [alanine])!
[Edited on 23-8-2006 by Nicodem]
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
Punk
Hazard to Self
Posts: 64
Registered: 22-2-2006
Member Is Offline
Mood: No Mood
|
|
???
Please explain how alanine can " act as an acid" if conditions were made basic with sodium bicarb and all solids were filtered out beforehand?
|
|
|
Nicodem
Super Moderator
Posts: 4230
Registered: 28-12-2004
Member Is Offline
Mood: No Mood
|
|
| Quote: | Originally posted by Punk
Please explain how alanine can " act as an acid" if conditions were made basic with sodium bicarb and all solids were filtered out beforehand?
|
I underlined the stage where you failed to see the point of the problem. Now, try again to carefully read what I wrote in my previous post and perhaps
you will be able to answer your question by yourself.
…there is a human touch of the cultist “believer” in every theorist that he must struggle against as being
unworthy of the scientist. Some of the greatest men of science have publicly repudiated a theory which earlier they hotly defended. In this lies their
scientific temper, not in the scientific defense of the theory. - Weston La Barre (Ghost Dance, 1972)
Read the The ScienceMadness Guidelines!
|
|
|
2bob
Harmless
Posts: 21
Registered: 29-8-2006
Member Is Offline
Mood: No Mood
|
|
Sorry to ask a dumb question, but did anybody think to add up the Hydrogen atoms?
It appears to me that C7H6O + C3H7N02 = C9H13NO + CO2, which would appear to be synomonous with PPA rather than anything you may actually want.
However, by performing a streckert degredation (dunno, how about CaOCl?) on Phenylalanine to get Phenylacetaldehyde (C8H8O) and then performing this
same reaction, it may be possible to come up with what is sought, eg.C10H15NO + CO2?
|
|
|
| Pages:
1
2
3
..
7 |











